There is emerging interest in cyclic conjugated molecules that happen to have triplet spin states and which might be expected to follow a 4n rule for aromaticity.[1] The simplest such system would be the triplet state of cyclobutadiene, for which a non or anti-aromatic singlet state is always found to be lower in energy. Here I explore some crystal structures containing this motif for possible insights.
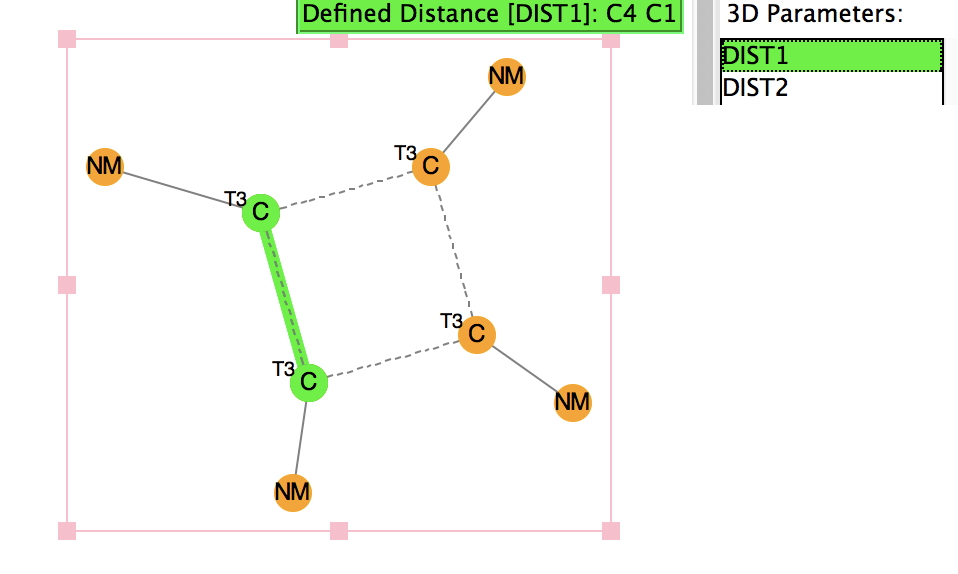
My search query is shown below, and the search is constrained so that the four substituents are Si, C or H.
The results show three clusters. The top left and bottom right have one long bond length ~1.6Å and the other much shorter at ~1.35Å (Δr ~0.25Å) The central region contains two examples, 2 where the difference between the two lengths is rather smaller and 1 where they are equal.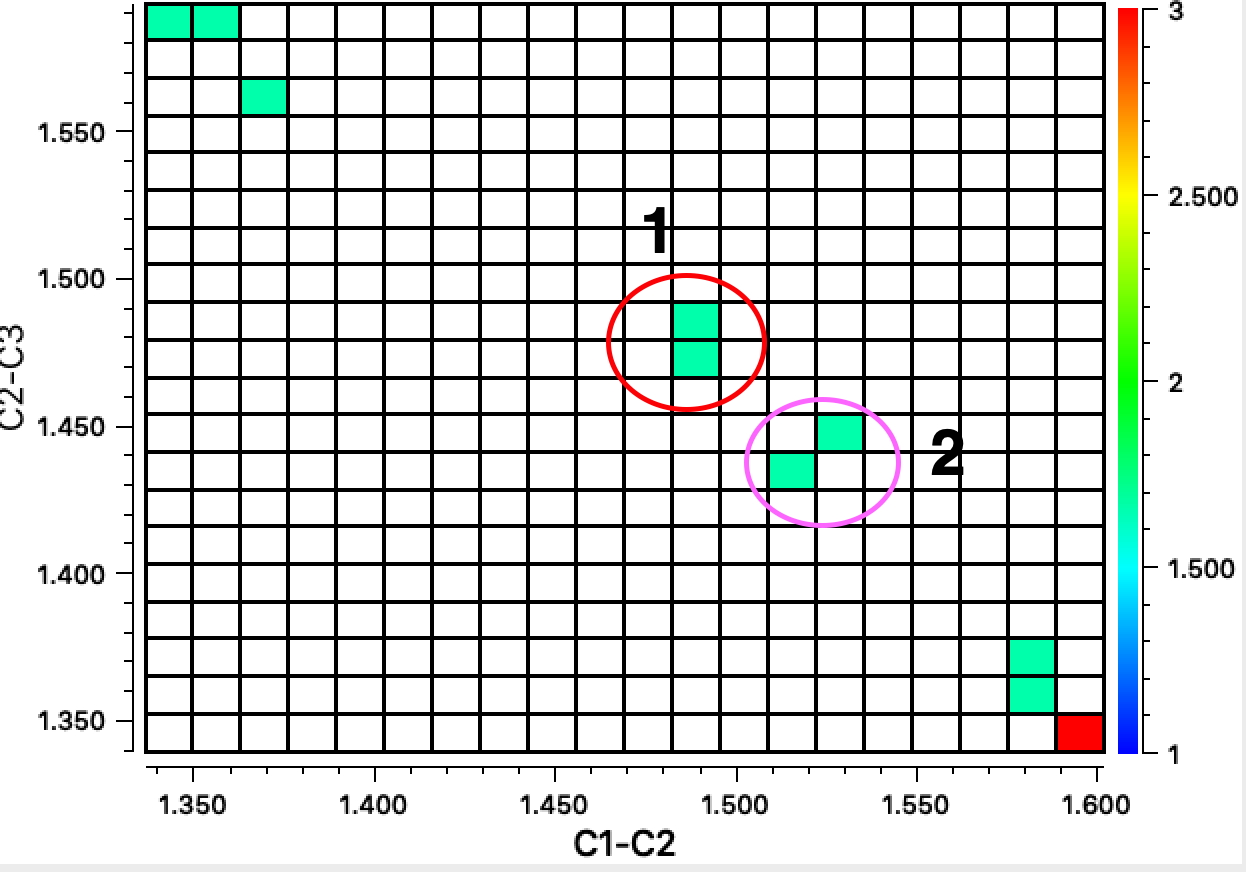
The first example 1[2] is in fact the di-anion of cyclobutadiene and as a 6π aromatic, one indeed expects the C-C bonds to be equal in length. The second 2 is tetra t-butylcyclobutadiene as reported in 1983.[3] At room temperature the two C-C bond lengths are 1.464 and 1.483Å, at -30°C, 1.466 and 1.492Å and at -150°C 1.441 and 1.526Å (Δr 0.085Å). These results led to the conclusion that this species was not intrinsically square but rectangular, as expected of singlet cyclobutadiene. The equalisation was attributed to equal populations of two disordered rectangular orientations averaging to an approximately square shape at higher temperatures.
But why is the behaviour of this particular cyclobutadiene different from the others in the plot above? Perhaps the answer lies these in the results of the Schreiner group[4], in which the dispersion attractions of substituents such as t-butyl can have substantial and often unexpected effects on the structures of molecules. So it is reasonable to pose the question; could the room temperature bond length differences of 2 be smaller compared with the other more extreme examples as a result of dispersion effects?
Here I have computed the singlet geometry of tetra t-butylcyclobutadiene at the B3LYP+D3BJ/Def2-TZVPP level (i.e. using the D3BJ dispersion correction, FAIR data DOI: 10.14469/hpc/4924). Δr for this singlet state is 0.264Å, larger than apparently from the crystal structure, but in agreement with the other crystal results as seen above.
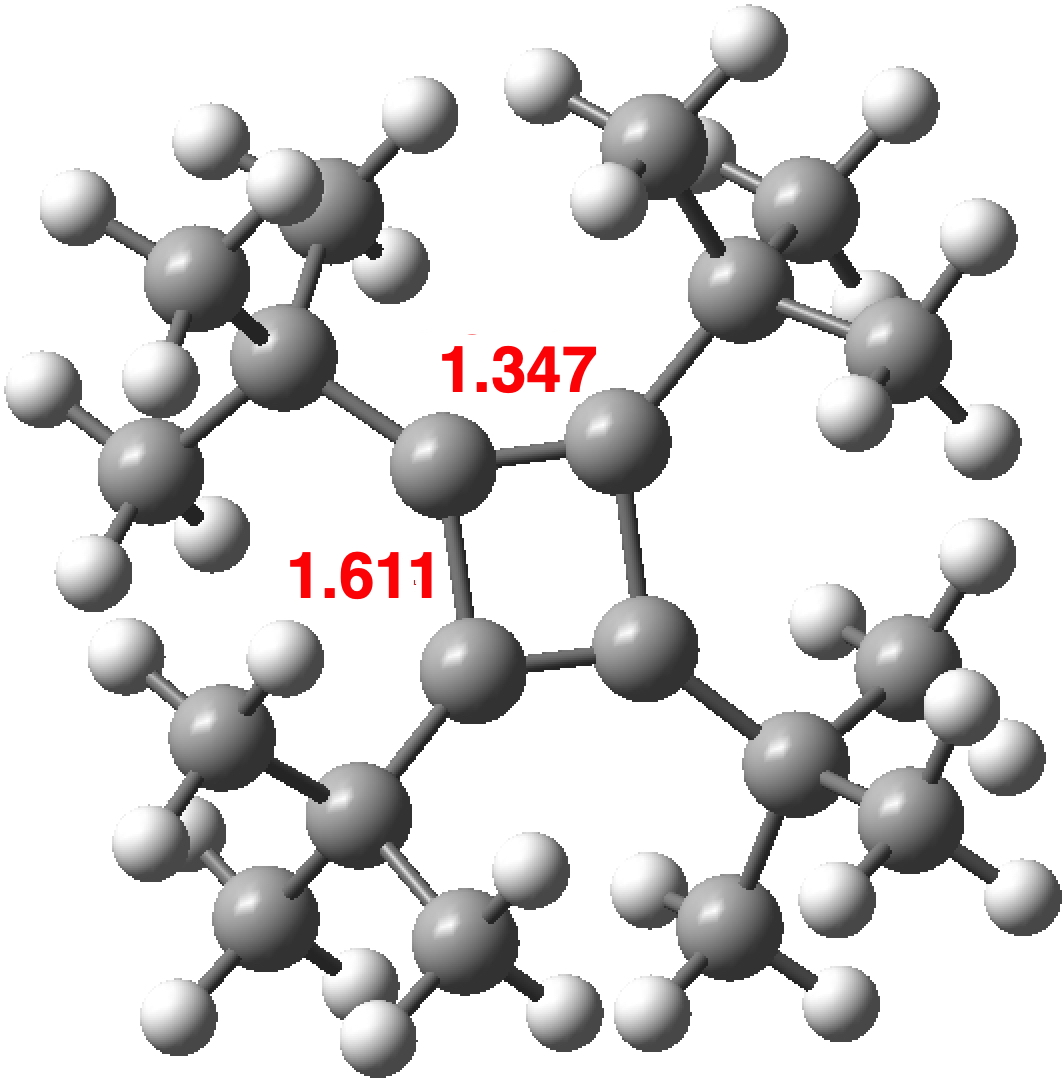
The origins of the measured structure of 2 must be in the barrier to the automerisation of the singlet state. For normal cyclobutadienes, this must be relatively high since the transition state is presumably anti-aromatic. High enough that the averaging of the two rectangular structures is slow enough that it manifests as two different bond lengths. But in 2, as the temperature of the crystal increases, the bonds become more equal, suggesting a lower barrier to the equalisation than the other examples. This is also supported by the apparent identification of a triplet square state for the tetra-TMS analogue of tetra-tert-butyl cyclobutadiene derivative [5] which again suggests that dispersion might favour a square form over the rectangular one.
To finish, I show the crystal structure search for the 8-ring homologue of cyclobutadiene, plotted for the two adjacent C-C lengths and (in colour) the dihedral angle associated with the three atoms involved and the fourth along the ring. Cluster 1 represents various boat-shaped derivatives with very different C-C bond lengths. Cluster 2 are all ionic, and as per above represent a planar 10π-electron ring. Cluster 3 are mostly “tethered” molecules in which additional rings enforce planarity.
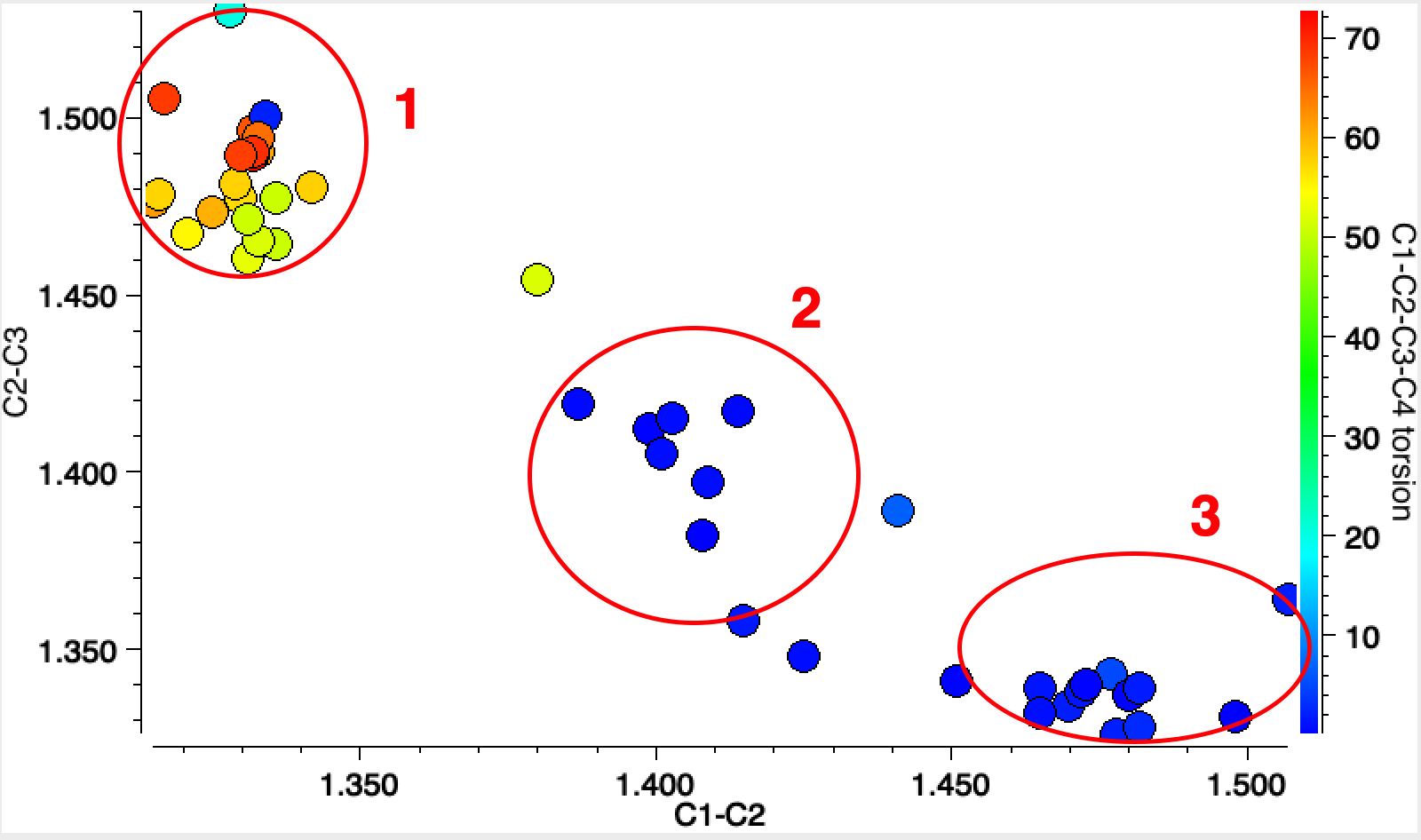
COT
Unfortunately, none of these derivatives include tert-butyl or TMS derivatives in adjacent positions around the central ring. Perhaps octa(t-Bu)cyclo-octatetraene or its TMS analogue would be interesting molecules to try to synthesize!
Author
References
- A. Kostenko, B. Tumanskii, Y. Kobayashi, M. Nakamoto, A. Sekiguchi, and Y. Apeloig, "Spectroscopic Observation of the Triplet Diradical State of a Cyclobutadiene", Angewandte Chemie International Edition, vol. 56, pp. 10183-10187, 2017. https://doi.org/10.1002/anie.201705228
- T. Matsuo, T. Mizue, and A. Sekiguchi, "Synthesis and Molecular Structure of a Dilithium Salt of the <i>cis</i>-Diphenylcyclobutadiene Dianion", Chemistry Letters, vol. 29, pp. 896-897, 2000. https://doi.org/10.1246/cl.2000.896
- H. Irngartinger, and M. Nixdorf, "Bonding Electron Density Distribution in Tetra‐<i>tert</i>‐butylcyclobutadiene— A Molecule with an Obviously Non‐Square Four‐Membered ring", Angewandte Chemie International Edition in English, vol. 22, pp. 403-404, 1983. https://doi.org/10.1002/anie.198304031
- S. Rösel, H. Quanz, C. Logemann, J. Becker, E. Mossou, L. Cañadillas-Delgado, E. Caldeweyher, S. Grimme, and P.R. Schreiner, "London Dispersion Enables the Shortest Intermolecular Hydrocarbon H···H Contact", Journal of the American Chemical Society, vol. 139, pp. 7428-7431, 2017. https://doi.org/10.1021/jacs.7b01879
- https://doi.org/
Tags: antiaromaticity, aromaticity, Baird's rule, Conjugated system, crystal structure search, energy, Hückel's rule, Nature, Physical organic chemistry, Physical sciences, search query, Triplet state