Another selection (based on my interests, I have to repeat) from WATOC 2017 in Munich.
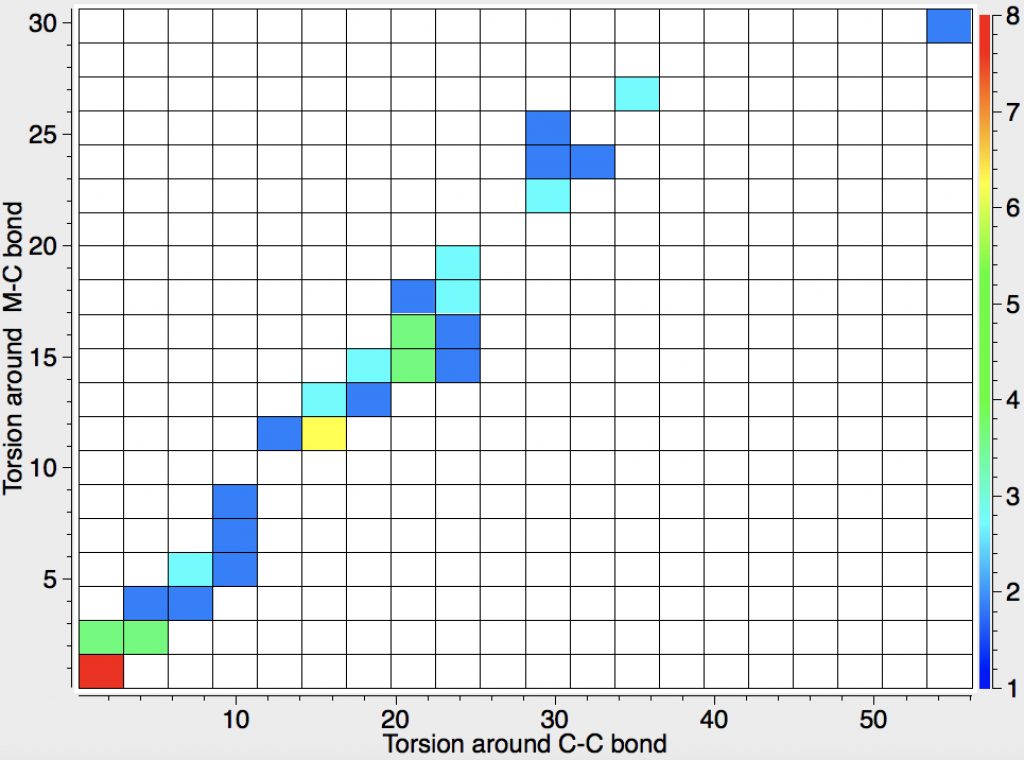
- Odile Eisenstein gave a talk about predicted 13C chemical shifts in transition metal (and often transient) complexes, with the focus on metallacyclobutanes. These calculations include full spin-orbit/relativistic corrections, essential when the carbon is attached to an even slightly relativistic element. She noted that the 13C shifts of the carbons attached to the metal fall into two camps, those with δ ~+80 ppm and those with values around -8 ppm. These clusters are associated with quite different reactivities, and also seem to cluster according to the planarity or non-planarity of the 4-membered ring. There followed some very nice orbital explanations which I cannot reproduce here because my note taking was incomplete, including discussion of the anisotropy of the solid state spectra. A fascinating story, which I add to here in a minor aspect. Here is a plot of the geometries of the 52 metallacyclobutanes found in the Cambridge structure database. The 4-ring can be twisted by up to 60° around either of the C-C bonds in the ring, and rather less about the M-C bonds. There is a clear cluster (red spot) for entirely flat rings, and perhaps another at around 20° for bent ones, but of interest is that it does form something of a continuum. What is needed is to correlate these geometries with the observed 13C chemical shifts to see if the two sets of clusters match. I include this here because in part such a search can be done in “real-time” whilst the speaker is presenting, and can then be offered as part of the discussion afterwards. It did not happen here because I was chairing the meeting, and hence concentrating entirely on proceedings!
- Stefan Grimme introduced his tight binding DFT method, an ultra fast procedure for computing large molecules and in passing noted the arrival of his D4 procedure (almost everyone currently uses D3 methods for this, including many of the results reported on this blog) for correcting for dispersion energies in molecules based on computed charge dependencies using the TBDFT methods. Thus we see dispersion as a property which is based on the wavefunction of the molecule, but still fast enough to accurately correct dispersion energies. He followed this with his automated procedures based on the TBDFT methods for computing full spin-spin coupled 1H NMR spectra of organic molecules. The core of this method is to recognise conformational and rotational freedoms and to compute the NMR properties for all identified isomers. These parameters are then Boltzmann averaged prior to computation of the final spin-coupled simulated frequency domain spectrum (rather than inverting this procedure by computing spin-coupled spectra of all rotamers and conformations and then averaging the spectral envelopes). This should widely revolutionise the interpretation of 1H NMR spectra by synthetic chemists.
- Another automated tool for synthetic chemists was presented by Jan Jenson, and can be seen here. It used MOPAC PM3 semi-empirical theory to compute relative proton affinities for a series of regioisomers as a prelude to predicting the position of aromatic electrophilic substitutions in heteroaromatic molecules. Try it out by putting a SMILES string into the box provided (e.g. COC1=CC=CC=C1) waiting a bit and seeing what the prediction is (it should be p- for the preceding example). During Q&A, a question was asked about the canonical “purity” of the SMILES (the one used in this tool comes from the Chemdraw program, which might not be identical to a SMILES for the same molecule produced by a different program), and whether an InChI descriptor might be better (also produced by Chemdraw, but perhaps a bit more canonical). Also asked was whether the prediction for an electrophile rather larger than a proton might not give good predictions? This one perhaps could be tested by readers, who could report back here?
- Walter Thiel completes the semi-empirical theme when he reported the new ODM2 method, the D now including dispersion. This is a powerful program, which includes e.g. full CI (configuration interaction + gradients) capability and is especially good for excited states, for dynamic simulations, and for combining these into dynamic photochemical simulations. This was applied to the chromophore in the famous “nanocar” in studying the dynamics of the photochemical rotation of the motor of the car (the thermally induced rotation was not studied). At the time that the nanocar caught my attention, I wondered about how the four independent molecular motors synchronised their rotations to allow the car to drive in a straight line. No doubt the answer is known, and if anyone reading this knows, please tell! It is probably a dynamics problem on four rotors (Walter reported just on one!).

Author
Tags: chemical shifts, Chemistry, City: Munich, Jan Jenson, metal fall, Munich, Odile Eisenstein, Quotation, speaker, Stefan Grimme, Transition metal, Walter Thiel, World Association of Theoretical and cOmputational Chemists
Unless I misunderstand the question, it seems to me that the Q. about the “canonical purity” of the SMILES is not well posed. What does it even mean? If two kits canonicalize anisole as COc1ccccc1 and COC1=CC=CC=C1, they are still encoding the same molecule, and if the second representation is considered non-aromatic by any piece of code, whichever part is doing the molecular recognition has a problem.
SMILES representations that differ in formal charge (e.g, the phenolate vs. the carbonyl representation of pyridone) will usually not canonicalize to the same representation, and indeed, at the molecular-mechanics level, might be recognized differently, which is a problem; but I would guess that even so, differences would not arise at the quantum level.
What I was getting at is that the toolkit was developed using SMILES generated by Chemdraw. If fed a SMILES generated using different codes, the toolkit might not necessarily generate the same molecule. I believe OpenSMILES is now available as the optimal SMILES definition (Daylight, the original creator of SMILES is no longer active), but whether Chemdraw might support it is unknown (at least by myself).
At very least the toolkit should also accept InChI.
We use RDKit to convert SMILES to coordinates, so it really comes down to how well RDKit handles non-canonical SMILES
Could you repeat why you think using Br+ as a probe instead of H+ could prove problematic (as you mentioned in our discussion)?
Yes, “really comes down to how well RDKit handles non-canonical SMILES”, and perhaps it handles it really well!
Re: Br(+), bromine often results in aggregated ions, particularly the anion. Should the anion be included in your calculations therefore? Is the unbalanced cation a good enough model (in steric systems) or should one strive to calculate the “Wheland” cation-anion pair? See DOI: 10.1002/jcc.23985 for more info.
It might be fun to use a large carbocationic replacement for H+ (t-butyl might suffer from methyl migrations); perhaps adamantanyl cation?
What Peter Shenkin said. Also, the questioner forgets that quantum chemical calculations are not invariant to atom order (as far as I know) but I bet the questioner does not canonicalise the atom order in their Gaussian files.
Also, no tool should ever accept InChI as input. It’s the one thing it’s not for. 90% of the time you get a different structure.
Just to clarify: quantum chemical calculations are not invariant to atom order, I think what was meant is that quantum chemical calculations are invariant to atom order (although the subsequent choice of coordinates for geometry optimization may influence the outcome slightly, or even a lot in pathological cases due to roundoff errors).
Re the use of InChI for input. It is true InChI was not designed to be reversible, but it seems to be very successful in practice if the molecule is not organometallic, polymeric or tautomeric. The debate continues as to whether it is just an Identifier or also a chemoinformatics tool.