The effects of loading up lots of dispersion attractions (between t-butyl groups) into a compact molecule has the interesting consequence of allowing two “non-bonded” hydrogen atoms to approach to ~1.5Å of each other, thus creating the appearance of a “bond” where one normally would not be found. Can such an effect be injected into other combinations of two atoms, say H and F? Here I briefly explore this notion.
The system is a slightly modified version of the one[1] already studied; R3C-F…H-CR3 (R=3,5-bis-t-butylphenyl), and a B3LYP+D3BJ/6-311G(d,p) calculation‡ (with C3-symmetry imposed) shows (DOI: 10.14469/hpc/2734) the following, with the key atom pair distances shown below. Note the abnormally short F…H distance, and the relatively long C-F one.
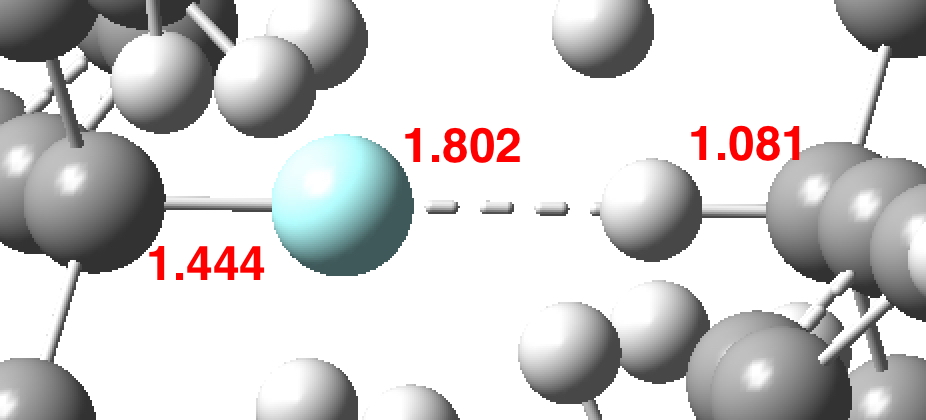
Note the casual phrase “C3-symmetry imposed”. This is a little “shortcut” one can try to use to shorten the calculation time. I should explain that on our computer system here, we are allowed a maximum of 72 hours per calculation. I already suspected that without the use of such symmetry the calculation would take longer and so used symmetry to “fit the calculation in” to this time slot. In the event it took 55 hours. There is a simple test however to see if this shortcut is justified; does the resulting molecule have 3N-6 real normal vibrational modes (i.e. ones with +ve force constants)? In fact this system fails this test; two of these modes have small negative force constants, corresponding to ν -11 cm-1. You might think this is small enough to perhaps attribute to e.g. the use of too-small a basis set or some other computational imperfection. Actually, although -11 cm-1 is numerically small, the mass-weighting associated with the vibration is in effect the entire system (below) and hence this mode is indeed significant.
So time to release the symmetry and when one does this an entirely different geometry emerges (DOI: 10.14469/hpc/2736) for which now all the 3N-6 normal modes have +ve force constants.
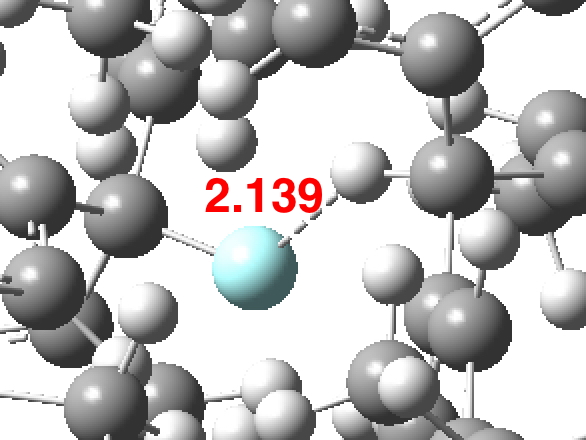
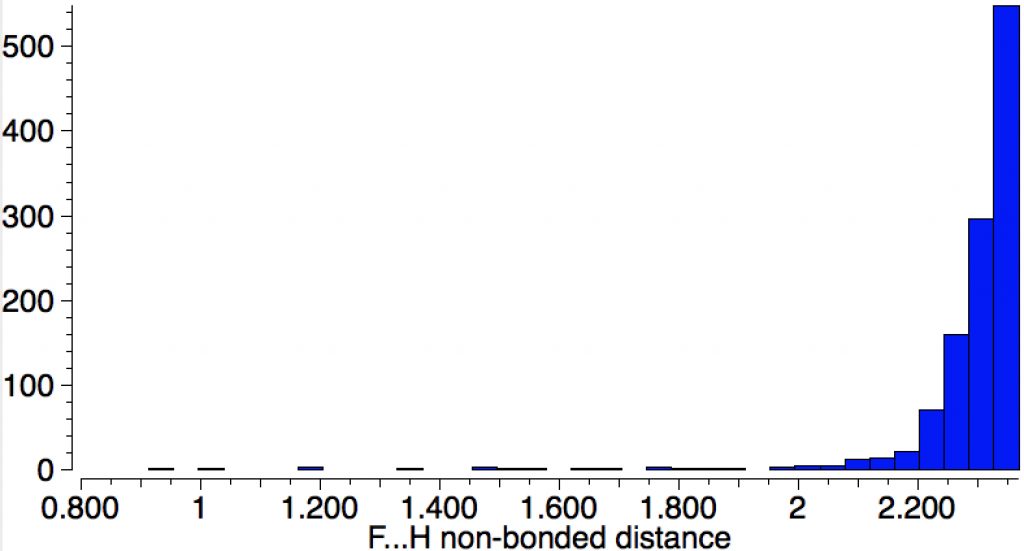
The “non-bonded” F…H interaction is now considerably longer, although still ~0.5Å shorter than the sum of the van der Waals radii (~2.65Å). This F…H non-bonded distance shows up as below in the Cambridge structure database (CSD) distribution. This suggests the shortest interaction is indeed ~2.1Å. The string of isolated examples with shorter distances down to < 1Å are very likely all crystallographic artefacts or errors.
So we may conclude that using the same system that was so successfully used to demonstrate the dispersion-induced ultra-short H…H distance cannot be modified to produce any such extreme effects in the F…H pair. Perhaps indeed “dispersion bonds” will always be limited to H…H pairs.
‡ When this method is used for the original H…H system, it yields a H…H distance of 1.529Å for which all the normal vibrational modes are real; DOI: 10.14469/hpc/2739).
Author
References
- S. Rösel, H. Quanz, C. Logemann, J. Becker, E. Mossou, L. Cañadillas-Delgado, E. Caldeweyher, S. Grimme, and P.R. Schreiner, "London Dispersion Enables the Shortest Intermolecular Hydrocarbon H···H Contact", Journal of the American Chemical Society, vol. 139, pp. 7428-7431, 2017. https://doi.org/10.1021/jacs.7b01879
Interesting test! Do you agree if we call these dispersion bonds “charge-shift bond”? There must be some elements of CS bonding in these bonds to stabilise them.
Cina, my intuition says that the H-H dispersion bonds will not be CSB’s, but I have no justification for that assumption.
I do think that such ‘dispersion-bonds’ will be limited to H-H interactions, though. Over the course of examining the ELF topology of various systems, it seems to me that hydrogen is unique in that it lacks an ELF attractor at the nucleus. Thus, the nucleus of an H atom can reside inside the ELF basin for its valence interactions, whereas all other elements (with the possible exception of He) exhibit distinct core and valence ELF basins. This has the effect of imposing a more more “ray-like” character (in the high school geometry sense) to the valence ELF basins of non-hydrogen elements.
I believe this is (at least part of the reason) why, for example, [diborane exhibits 3c2e bonding, whereas Al2Cl6 does not](https://chemistry.stackexchange.com/a/64748/11367). Further, based also on this linked analysis, while Al2Cl6 does appear to have some interesting hypovalent resonance phenomena going on, the electron density at the Al-Cl LCPs (~0.048) is low enough that the bonds appear much more like true ionic bonds, than charge-shift bonds. (For comparison, I calculate the density at the LCP of NaF, a true ionic species, to be ~0.055.)
Although, now that I look at the banana bond LCPs in the ELF topology for diborane, they actually do exhibit characteristics rather like charge-shift bonds. I rescind the assertion of the first paragraph of my initial comment.
I really need to learn a VB software package, so that I can solidly confirm whether the inferences about charge-shift character I’m trying to draw from QTAIM/ELF analysis are actually correct or not!