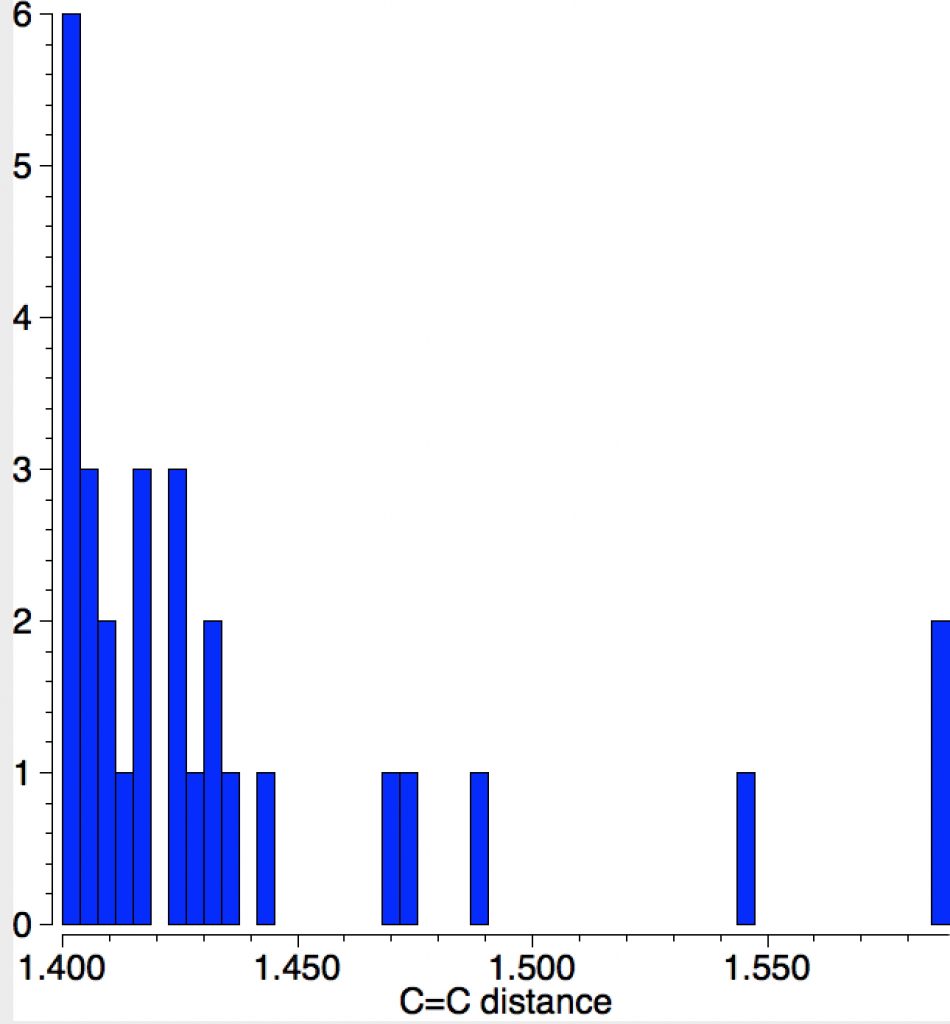
Following on from a search for long C-C bonds, here is the same repeated for C=C double bonds.
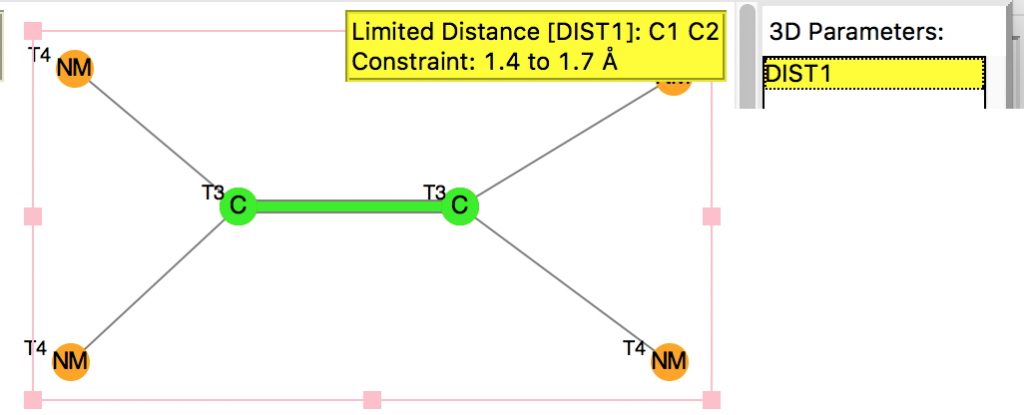
The query restricts the search to each carbon having just two non-metallic substituents. To avoid conjugation with these, they each are 4-coordinated; the carbons themselves are three-coordinated. Further constraints are the usual no disorder, no errors and R < 0.1 and the C=C distance > 1.4Å (the standard value is ~1.32-1.34Å). The search query is deposited as DOI: 10.14469/hpc/1959[1]
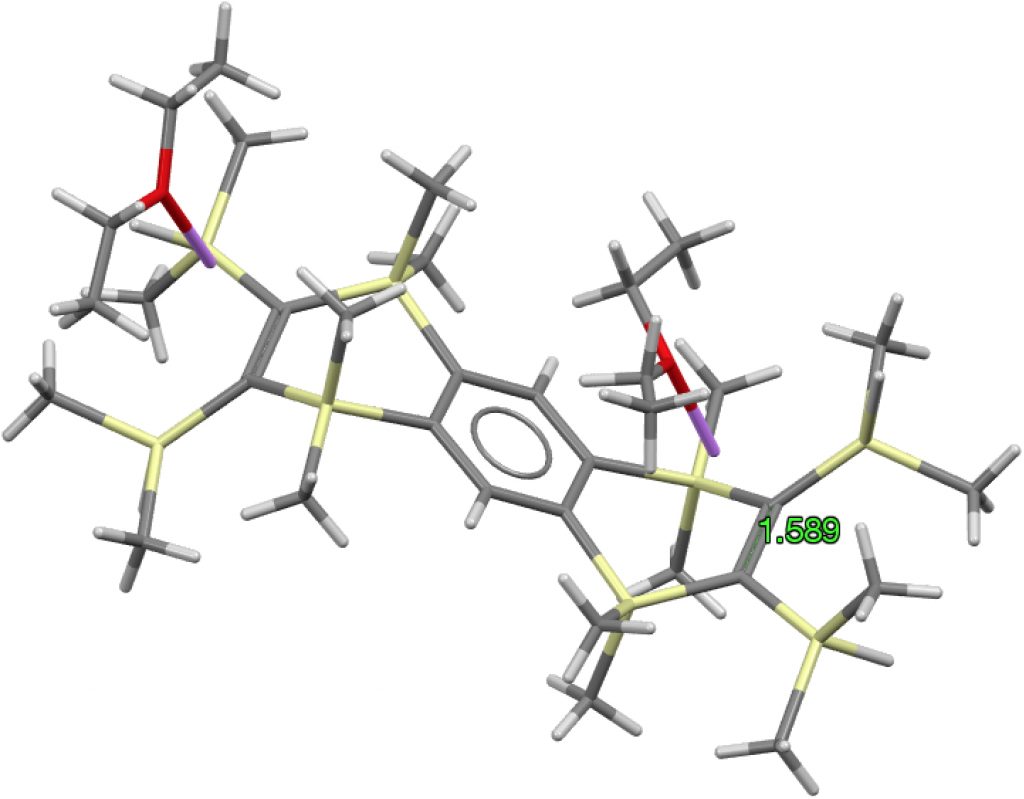
The apparent longest example is LIRVEN, DOI: 10.5517/CC4R2MK[2] with a value of 1.589Å, longer than most C-C single bonds! Closer inspection reveals the presence of lithium cations, and so the molecule bearing the C=C bond must sustain two negative charges. So this apparent C=C bond is in fact anionic, with one electron going into each of the π* orbitals, thus lengthening the CC bond.‡ Not a true example of a neutral C=C bond[3] but it now becomes interesting for what its spin state might be. Is it a biradical or a triplet for example? One to be investigated further I fancy! Another example of this type is QUKCEE[4]
This next FAZWIM has a C=C length of 1.546Å. It is an old structure (1986), and comes without attached hydrogen atoms. Although drawn with no hydrogens on the central C=C bond, the length suggests this molecule is simply mis-assigned.†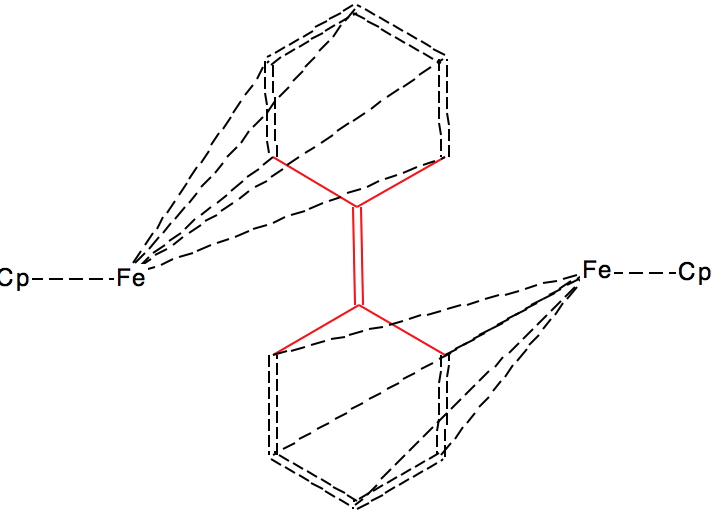

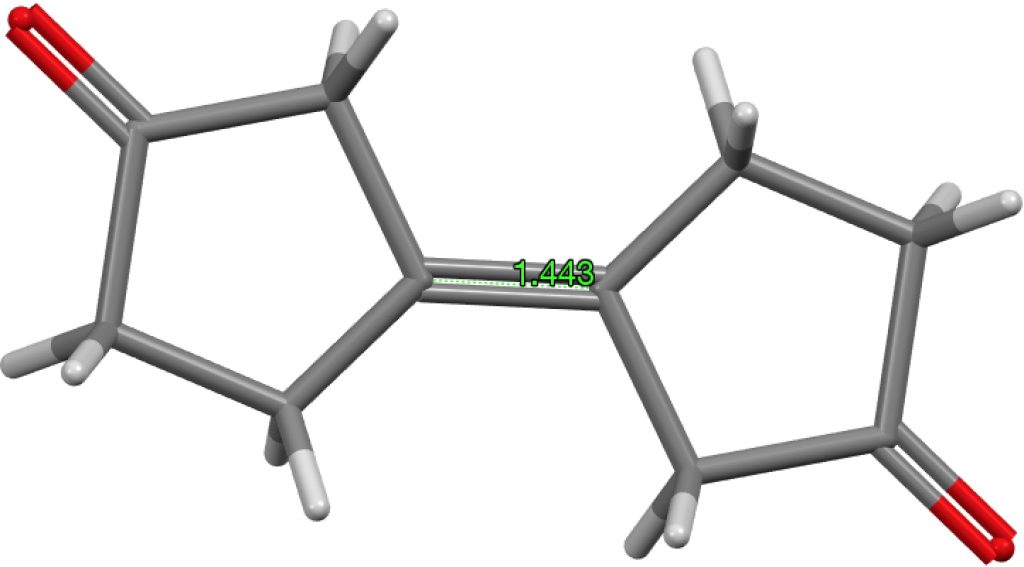
The final example I will highlight is pretty ordinary looking and published in 2016 as a private communication; ALOVOO, DOI: 10.5517/CCDC.CSD.CC1LJSWS[5] with a C=C length of 1.443Å. Again no obvious reason for the bond to be longer than normal.‡†
In hunting for such unusual deviations from the norm, the most obvious explanation is normally some anomaly in the crystallographic analysis. Although the CSD (crystal structure database) is a very heavily curated resource, it seems unlikely that each deposition would be carefully inspected for its chemistry, and this must be our task here. But such anomalies can themselves point to interesting or unusual chemistry, which in turn can be subjected to quantum computation to see if either the unusual value can be replicated or other reasons identified. In this case, this exercise can been conducted by a human, but one can easily envisage the entire process being automated on a far larger scale. The future?
‡ In fact the stoichiometry shows each “double bond” is actually a di-anion, with two electrons entering each of the the π* orbitals.
†A calculation on the singlet state for the structure as drawn (ωB97XD/Def2-TZVPP, DOI: 10.14469/hpc/1960) gives a bond length of 1.342Å, i.e. that expected for a double bond. The triplet state is similar in energy, but with a much longer central bond length of 1.476Å, DOI: 10.14469/hpc/1962 but the geometry at the carbons is planar and not bent as shown above. The quintet state is 1.45Å and is again planar, doi 10.14469/hpc/1963. So calculations on FAZWIM strongly suggest the structure as shown is an error.
‡†The computed value is 1.324Å, perfectly normal. DOI: 10.14469/hpc/1966[6]
Author
References
- H. Rzepa, "Long C=C bonds", 2016. https://doi.org/10.14469/hpc/1959
- Matsuo, T.., Watanabe, H.., Ichinohe, M.., and Sekiguchi, A.., "CCDC 141348: Experimental Crystal Structure Determination", 2000. https://doi.org/10.5517/cc4r2mk
- T. Matsuo, H. Watanabe, M. Ichinohe, and A. Sekiguchi, "Reduction of the 1,4,5,8-tetrasila-1,4,5,8-tetrahydroanthracene derivative with lithium metal. Isolation and characterization of the tetralithium salt of a tetraanion, and observation of an Si–H⋯Li+ interaction", Inorganic Chemistry Communications, vol. 2, pp. 510-512, 1999. https://doi.org/10.1016/s1387-7003(99)00136-7
- T. Matsuo, H. Watanabe, and A. Sekiguchi, "A Novel Tetralithium Salt of a Tetraanion and a Dilithium Salt of a Dianion, Formed by the Reduction of the Tetrasilylethylene Moiety. Synthesis, Characterization, and Observation of an Si-H···Li+ Interaction", Bulletin of the Chemical Society of Japan, vol. 73, pp. 1461-1467, 2000. https://doi.org/10.1246/bcsj.73.1461
- M.E. Light, S. Bain, and J. Kilburn, "CCDC 1475906: Experimental Crystal Structure Determination", 2016. https://doi.org/10.5517/ccdc.csd.cc1ljsws
- H. Rzepa, "ALOVOO", 2016. https://doi.org/10.14469/hpc/1966
Tags: Chemical bond, chemical bonding, Chemical nomenclature, Chemistry, Conjugated system, double bond, energy, Nature, Nonmetal, Organic chemistry, Physical organic chemistry, search query, Substituent
Since this post appeared on December 1st, I have been contacted regarding all these structures.
CCDC have kindly provided some responses.
1. FAZWIM was a simple error. But correcting it required “re-reading the paper. it does appear to reasonably clearly state that the benzene rings are eta-5 bonded and have their full complement of 6 hydrogen atoms”.
2. LIRVEN and QUKCEE again involves a detailed reading and analysis of the reporting article. Again, “I think that a better representation than what we have currently would be one with the negative charges on the carbon atoms with a single bond between them, as you noted. I have made these changes and they will be available to view very shortly”.
This is very much the 80:20 (or perhaps the 99.9:0.1) issue that automated checking procedures (Decifer) can only check so much, and that a human has to complete the analysis. Sometimes the level of analysis required is so deep and subtle that a human with limited time for each case cannot be expected to trap all such instances.
I have also been contacted about ALOVOO, more of which later.
A chance to hold my hands up and say, “CCDC entry ALOVOO in its original form was simply wrong!” This highlights two valid scientific points; the first being the usefulness of investigating outliers with apparent unusual properties, and the second, why was it wrong – are there lessons to learn?
The atoms either side of the long ‘double’ bond are actually disordered over two positions allowing for a much more normal sp3 geometry. This was obvious to me on a second look, but for some reason was not originally. This may have been due to distractions on the day, or more likely, I was overly influenced by what the chemist was expecting and duly obliged! I will be the first to admit this should never have happened, but with so many structures spanning so many years – the odd mistake will creep through.
So thanks to Henry for highlighting this error and giving me the chance to correct it. Maybe I can use it as a structure interpretation/validation exercise for the students.
NB. A corrected version has been submitted to CCDC.