Compounds with O-O bonds often have weird properties. For example, artemisinin, which has some fascinating stereoelectronics. Here is another such, recently in the news and known as HMTD (hexamethylene triperoxide diamine). The crystal structure was reported some time ago[1] and the article included an inspection of the computed wavefunction. However this did not look at the potential stereoelectronics in this species, which I now address here.
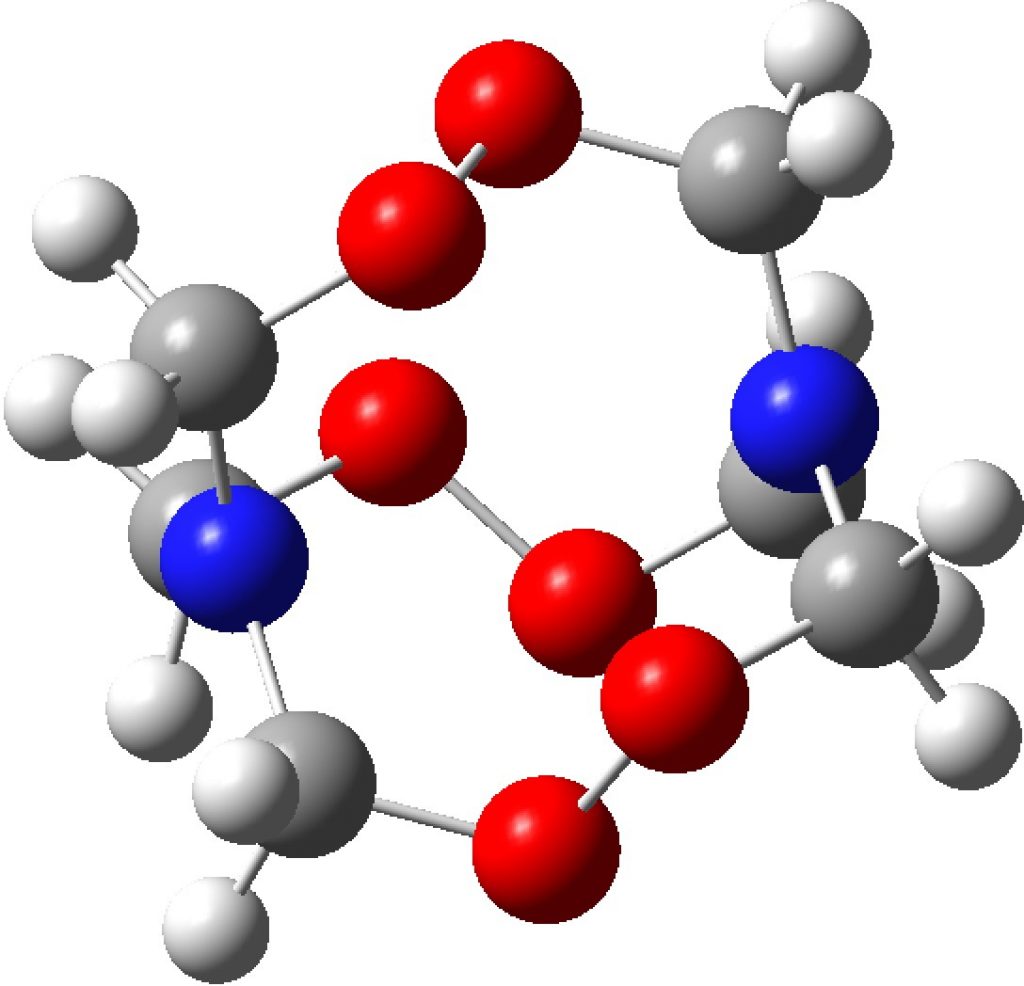
A ωB97XD/Def2-TZVPP calculation[2] can be analysed for the NBO-derived interaction terms. This identifies an electron donor (normally a bond or a lone pair) and its E(2) perturbation energy interaction with an acceptor (normally an empty σ* antibond). Here we are interested in the interaction between the nitrogen “lone pair” and the adjacent C-O σ* antibond, of which there are six in the molecule due to the D3 symmetry. E(2) is ~22.4 kcal/mol, which is a large effect (the equivalent value for the so-called anomeric interaction between an oxygen lone pair and a C-O antibond is ~18 kcal/mol). The effect of donation into the empty C-O σ* antibond is to weaken it, unless the effect is balanced by a reciprocal interaction in the opposing direction, which is often the case in sugar-derived anomeric effects. Sugars of course are thermally relatively stable. In the case of HMTD, the reverse effect would be an oxygen Lp donating into the N-C σ* antibond and this has the value of 14.5 kcal/mol. Since the two are not balanced, this presumably contributes to the very unstable nature of this molecule.
An alternative way of looking at what the electrons are up to is ELF, a function based on the electron density which identifies the centroids of electron basins. The red arrows point to the four basins associated with the nitrogen “lone pair” (mostly the dumb-bell-shaped p-atomic orbital, hence four basins), and the integration being 3.2e for each nitrogen. This is a rather odd number for a “lone pair”. There is undoubtedly something unusual about this wavefunction which has yet to be identified.
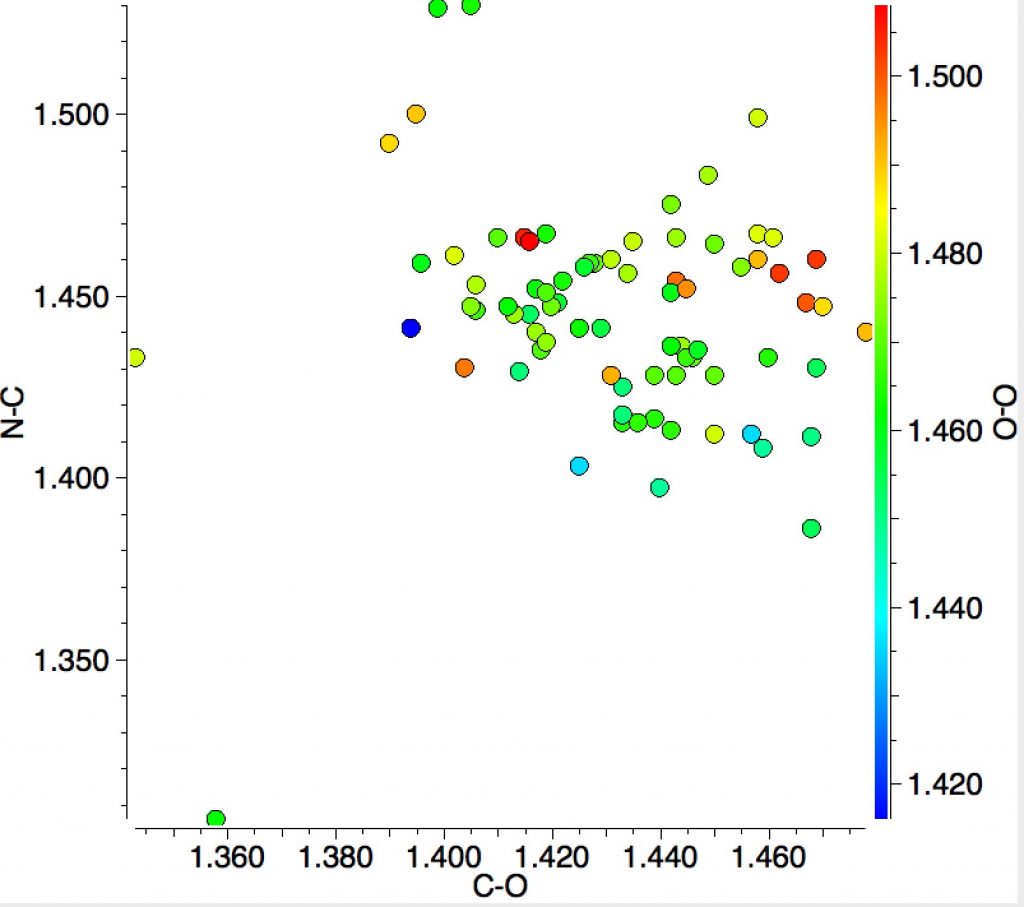
Finally, I ask how common the N(sp3)-C(sp3)-O-O structure motif might be? In fact the Cambridge structure database has 81 entries! The scatterplot below includes 51 of them (no disorder, no errors, R<0.05). No clear-cut conclusions emerge from these statistics, except just a hint that as the C-O distance gets longer, the N-C distance might get shorter and that shorter N-C lengths are associated with shorter O-O lengths.
Author
References
- A. Wierzbicki, E.A. Salter, E.A. Cioffi, and E.D. Stevens, "Density Functional Theory and X-ray Investigations of P- and M-Hexamethylene Triperoxide Diamine and Its Dialdehyde Derivative", The Journal of Physical Chemistry A, vol. 105, pp. 8763-8768, 2001. https://doi.org/10.1021/jp0123841
- H. Rzepa, "HMTD Hexamethylenetriperoxidediamine D3 NBO", 2016. https://doi.org/10.14469/hpc/1663
Tags: Amines, Artemisinin, Chemistry, Functional groups, Hexamethylene triperoxide diamine, Organic chemistry, Organic peroxides, Peroxide, perturbation energy interaction, Stereoelectronics
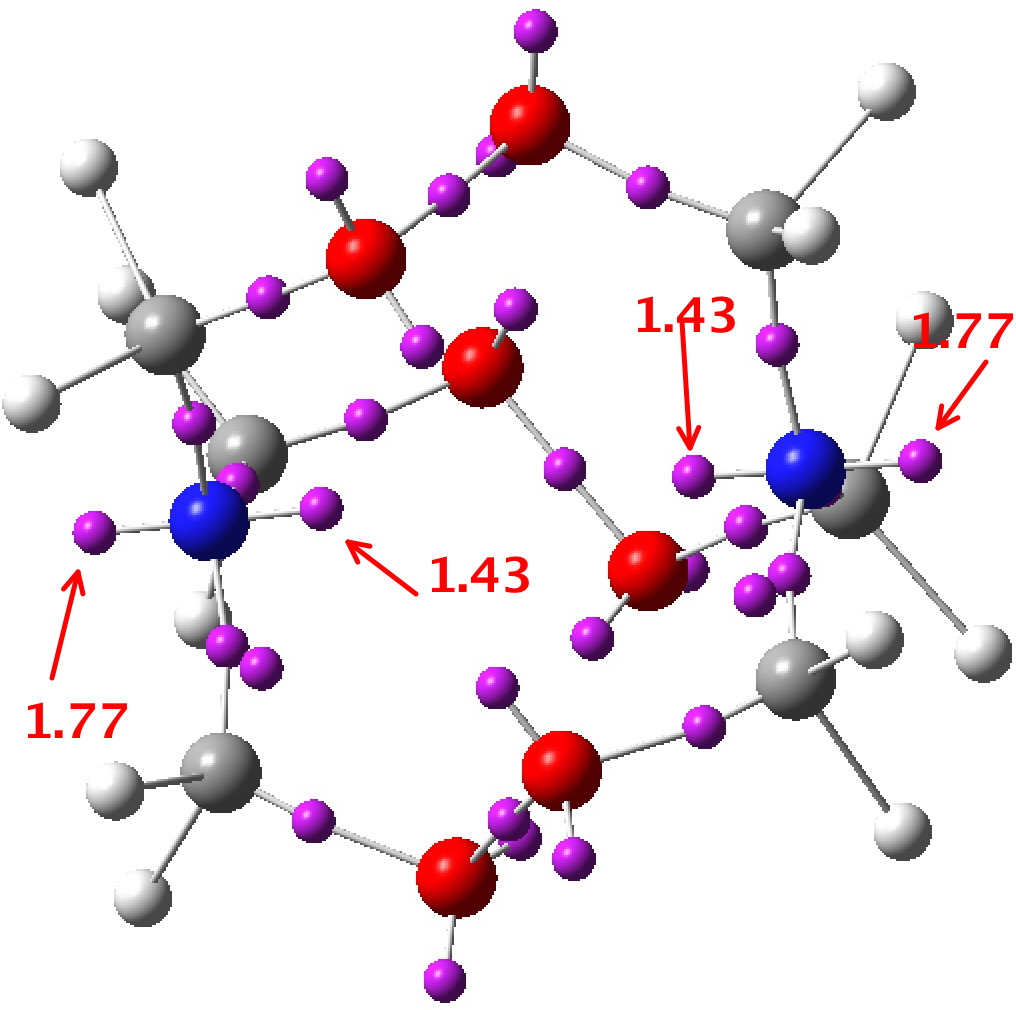
Here are some Wiberg bond orders for this system. The N-C is 1.006 distributed across six such bonds. This is slightly greater than 1.0, and indicates donation of the nitrogen lone pair into the C-N region. The geminal C-O bond orders are 0.938, significantly weakened from a normal C-O bond. The O-O bond order is 0.963.
I also include a rendering of the NBO nitrogen orbital. In fact, in the NBO localisation, it is NOT described as “Lp”, but as e.g. “BD ( 2) N 3 – C 7” which is used for a two-centre bond! There is sufficient donation of that electron pair to deserve the description of a two-centre interaction!