I previously used data mining of crystal structures to explore the directing influence of substituents on aromatic and heteroaromatic rings. Here I explore, quite literally, a different angle to the hydrogen bonding interactions between a benzene ring and OH or NH groups.
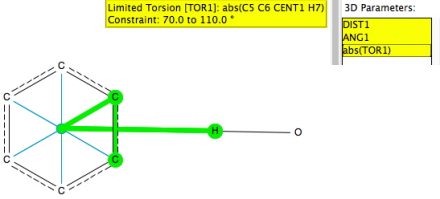
I start by defining a benzene ring with a centroid. The distance is from that centroid to the H atom of an OH or NH group and the angle is C-centroid-H. To limit the search to approach of the OH or NH group more or less orthogonal to the ring, the absolute value of the torsion between the centroid-H vector and the ring C-C vector is constrained to lie between 70-100° (the other constraints being no disorder, no errors, T < 140K and R < 0.05).[1]
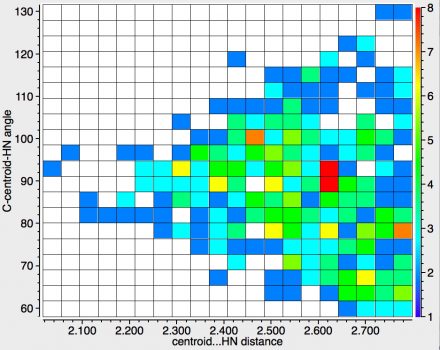
The above shows the results for NH groups interacting with the aromatic ring. The maximum distance 2.8Å is more or less the van der Waals contact distance between a hydrogen and a carbon and as you can see the contacts "funnel down" to the centroid at < 2.1Å. The shortest distance[2] is for ammonium tetraphenylborate, which you can view in e.g. spacefill mode here[3]
The other interesting close contact derives from a protonated pyridine[4], which can in turn be viewed here.[5] The main message from the distribution shown above is that as the distances between the HN and the centroid get shorter, the "trajectory" of approach remains orthogonal to the ring (the angle defined above remains ~90°) and heads towards the centroid of the π-cloud. The hotspot itself (red, ~2.6Å) also lies along this trajectory.
Recollect that when I used such hydrogen bonding to see if crystal structures discriminate between the ortho or meta positions of a ring carrying an electron donating substituent, it was the distance from a HO to the carbon that was measured as the discriminator. So it's a faint surprise to find that with HN, and without the necessary perturbation of an electron donating substituent, the intrinsic preference seems to be for the ring centroid and not any specific carbon atom of the ring.
So how about the OH group? There are in fact rather fewer examples, and so the statistics are a bit less clear-cut. But there is a tantalising suggestion that this time, the trajectory is not ~90° but rather less, implying that the destination is no longer the centroid of the π-cloud but one of the carbon atoms of the ring itself. For those who like to "read between the lines" and spot things that are absent rather than present, you may have asked yourself why I did not use NH probes in my earlier post. Well, it appears that the NH group is less effective at e.g. o/p discrimination than is an OH group.
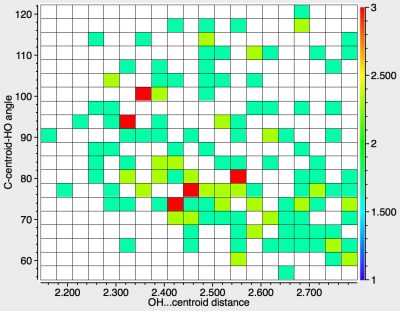
I can only speculate as to the origins (real or not) of the difference in behaviour between OH and NH groups towards a phenyl π-face. Perhaps it is simply bias in the CSD database? Or might there be electronic origins? Time to end with that phrase "watch this space".
Author
References
- H. Rzepa, "How does an OH or NH group approach an aromatic ring to hydrogen bond with its Ï-face?", 2016. https://doi.org/10.14469/hpc/673
- T. Steiner, and S.A. Mason, "Short N<sup>+</sup>—H...Ph hydrogen bonds in ammonium tetraphenylborate characterized by neutron diffraction", Acta Crystallographica Section B Structural Science, vol. 56, pp. 254-260, 2000. https://doi.org/10.1107/s0108768199012318
- Steiner, T.., and Mason, S.A.., "CCDC 144361: Experimental Crystal Structure Determination", 2000. https://doi.org/10.5517/cc4v6tz
- O. Danylyuk, B. Leśniewska, K. Suwinska, N. Matoussi, and A.W. Coleman, "Structural Diversity in the Crystalline Complexes of <i>para</i>-Sulfonato-calix[4]arene with Bipyridinium Derivatives", Crystal Growth & Design, vol. 10, pp. 4542-4549, 2010. https://doi.org/10.1021/cg100831c
- Danylyuk, O.., Lesniewska, B.., Suwinska, K.., Matoussi, N.., and Coleman, A.W.., "CCDC 819118: Experimental Crystal Structure Determination", 2011. https://doi.org/10.5517/ccwhc5w
Tags: 10.1021, 10.1107, 10.5517, aromaticity, benzene, Centroid, chemical bonding, data mining, Functional groups, Hydrogen bond, Physical organic chemistry, Pyridine, Simple aromatic rings, Supramolecular chemistry
How does a CH group approach an aromatic ring to hydrogen bond with its π-face? could be a follow up to the post above. Here are some crystal searches in which the ring-H-C interaction is split into H-C(sp), H-C(sp2), H-C(sp3). The shortest interactions to the ring centroid are again ~2.2Å and the trajectories for the last two types of H-C are both perpendicular to the plane of the aromatic ring, just like the HN in the post.
Prof. Rzepa,
Can you provide a recent literature reference for N-H..pi interactions? I see some theoretical and data-mining articles from 1998 to 2002, or so, but wondered if you know of something more recent. I and a colleague are working on a transition metal complex that shows this interaction, and we are seeking relevant recent comparisons.
Thanks
I have not tried to track down any reviews, but the CSD itself can provide some breakdowns. Here, I have searched for close contacts between the centroid of either a C=C or a C≡C bond and the H of an HN. The table shows the closest such contacts up to 2.3Å.
Using the Name (RefCode), you can then do individual searches for each entry, ie https://www.ccdc.cam.ac.uk/structures/Search?Ccdcid=KESDAO for one interesting example. This should then give the original literature reference for the entry.
Hope this helps.