Derek Lowe has a recent post entitled "Another Funny-Looking Structure Comes Through". He cites a recent medchem article[1] in which the following acetal sub-structure appears in a promising drug candidate (blue component below). His point is that orally taken drugs have to survive acid (green below) encountered in the stomach, and acetals are famously sensitive to hydrolysis (red below). But if X=NH2, compound "G-5555" is apparently stable to acids.[1] So I pose the question here; why?

This reminded me of some work we did a few years ago on herbicides containing such an acetal substructure, where one diastereoisomer was very unstable to hydrolysis (and hence did not have the lifetime required of a herbicide) whereas the other diastereomer was far less labile and hence more suitable.[2],[3] Crystal structures (below) revealed that the two C-O bond lengths of the labile form were very unequal in length (Δ0.043Å), whereas the stable form had two equal C-O lengths (1.40Å, Δ=0.0Å).
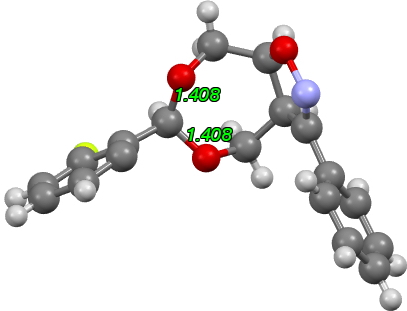
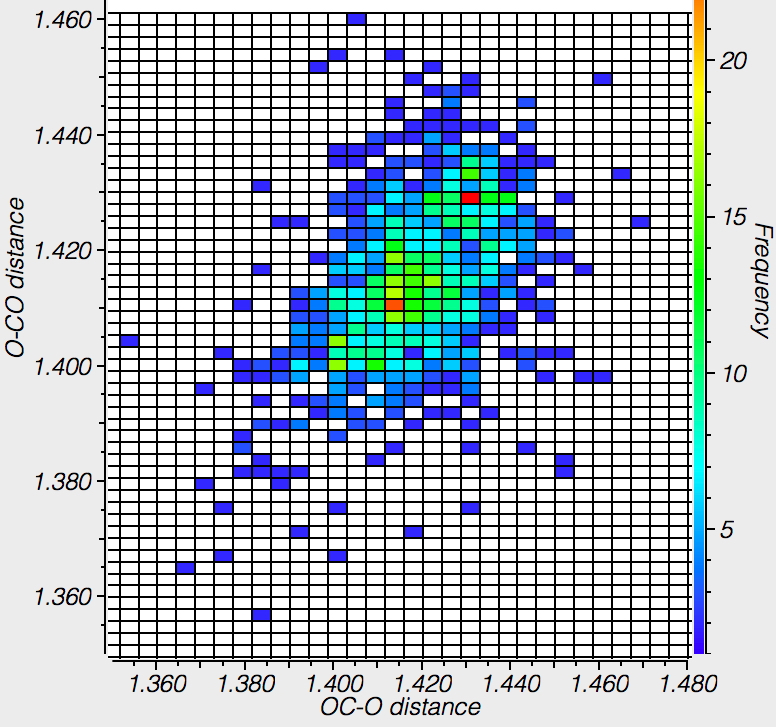
There is one example[4],[5] of an orthoformate in which the group equivalent to X is protonated as Me2NH+. For this example, all three C-O lengths are shorter than even the hydrolytically stable herbicide above (1.405, 1.402, 1.396Å). The distribution for 6-ring acetals in general shows hot-spots at ~1.415Å and 1.43Å (but sadly it is not possible to e.g. use this database to correlate these lengths with the aqueous stability of the entries).
Is this tentative further evidence that a group X = NH2 positioned as above in an acetal can inhibit its hydrolysis?
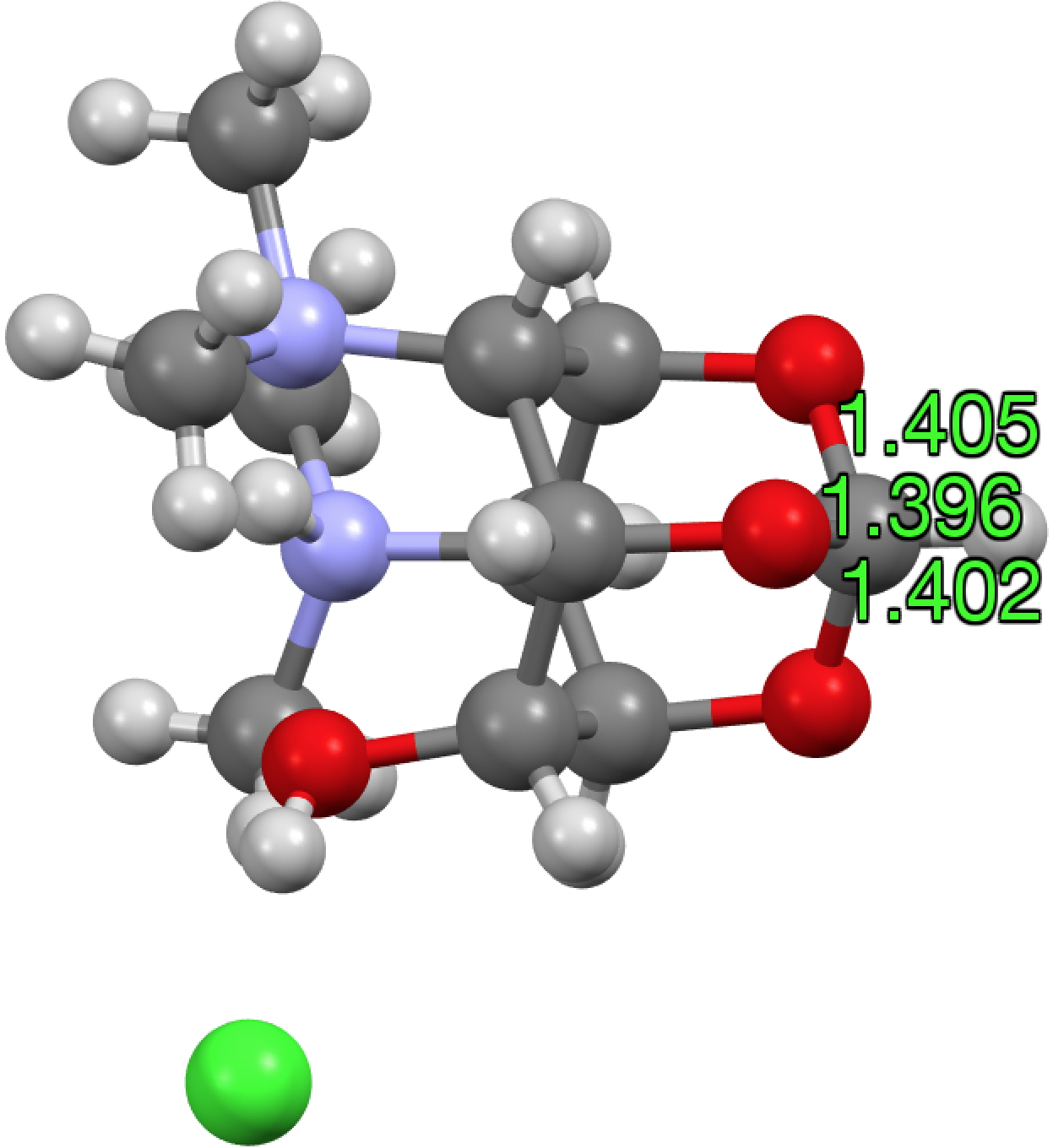
HUZKEZ, click for 3D
Time for calculations. A model (X=R=H) for the hydrolysis was constructed as above in which proton transfer from an acid (ethanoic) is achieved via a cyclic 8-ring transition state and which includes a continuum solvent field as ωB97XD/6-311G(d,p)/SCRF=water and one explicit water in the proton relay. The IRC looks thus:

This shows that the first event is protonation of an oxygen, closely followed by cleavage of the associated C-O bond, and ending with deprotonation of the erstwhile water molecule.
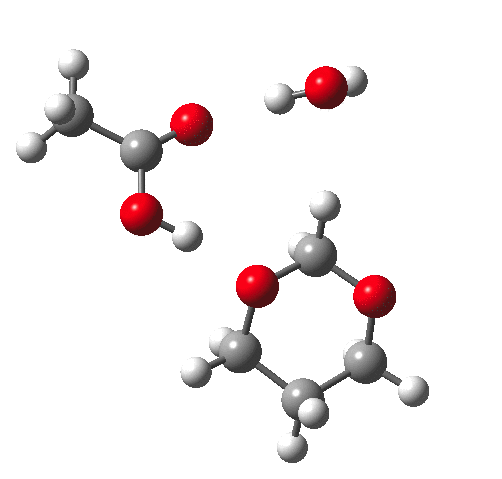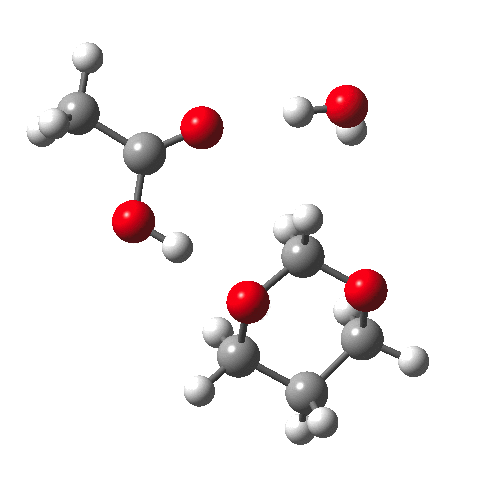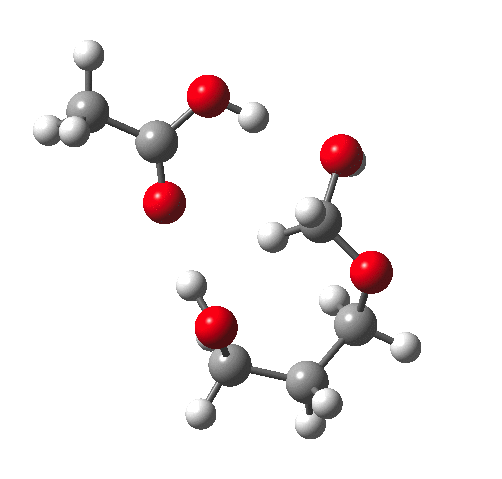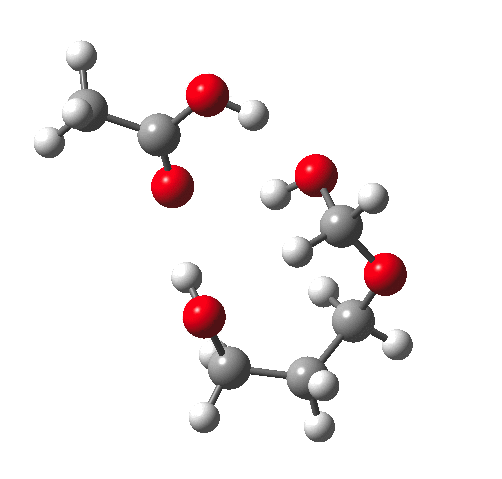
The value of ΔG‡298 is 38.2 kcal/mol (38.4 in relative total energy). Although rather high for a facile thermal reaction (perhaps the 8-ring TS is a bit too strained; possibly adding a second active water molecule to form a 10-ring might lead to a lower barrier?), we are more interested in the effect upon this barrier of group X (Table below).
| X | ΔE | ΔG‡ | DataDOI,TS | DataDOI,IRC |
|---|---|---|---|---|
| H | 38.4 | 38.2 | [6] | [7] |
| NH2,eq | 39.8 | 38.8 | [8] | [9] |
| NH3.Cl,eq | 45.1 | 43.1 | [10] | [11] |
| NH3.Cl,ax | 42.6 | 41.5 | [12] | [13] |
| CF3,eq‡ | 41.9 | 40.1 | [14] | [15] |
| SF5,eq‡ | 43.6 | 42.4 | [16] | [17] |
Introduction of X=NH3+.Cl– into an (equatorial) position which is antiperiplanar to the C-O bonds of the acetal produces a modified IRC profile. The barrier measured at a point IRC = -10 is ~41 kcal/mol, which is noticeably higher than for X=H. In fact the final barrier is even higher, since the reactant goes on to form a hydrogen bond between the water molecule and the Cl–, an extra stabilisation not present with X=H (and so not really appropriate to include in the comparison).

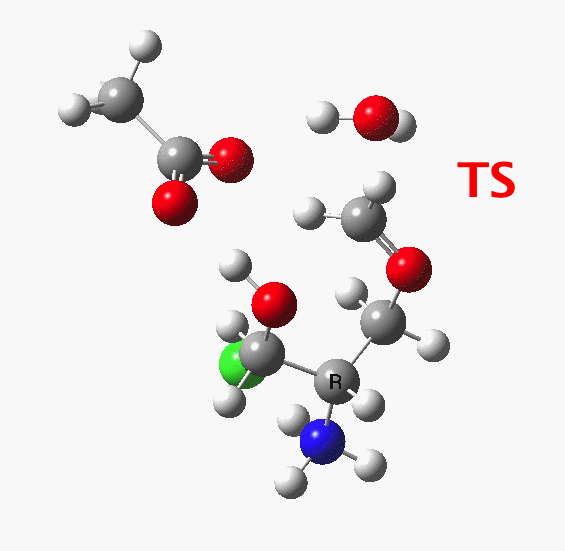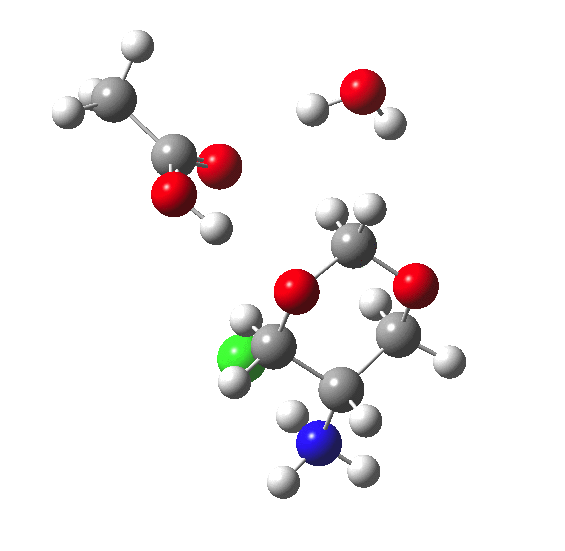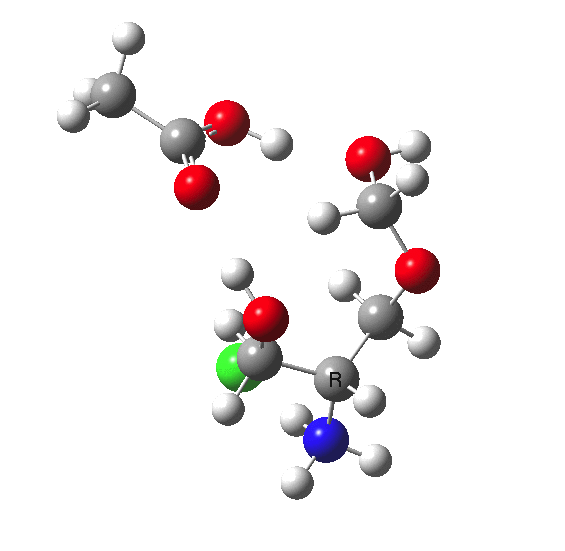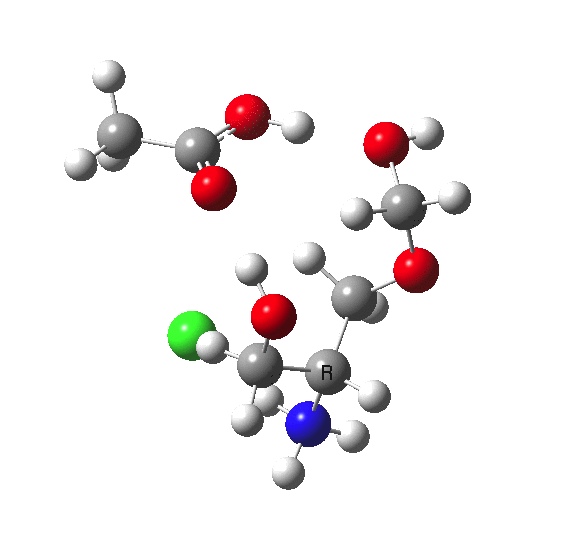
Placing the X=NH3+.Cl– into an (axial) position which is not antiperiplanar to the C-O bonds shows a lower barrier compared to the equatorial isomer. This difference can also be illustrated by the NBO localised orbital energies of the two reactants. With X=NH3+.Cl– axial, the lone pair on the oxygen being protonated by the acid has an energy of -0.464 au, whereas the equatorial equivalent is a "less reactive" -0.471 au (a difference in energy of 4.4 kcal/mol, which is VERY approximately related to the effects being discussed). I conclude that the inhibition of acetal solvolysis is induced by the presence of an electron withdrawing group X, via antiperiplanar effects on the basicity of the acetal oxygen. In moderately low pH, X=NH2 is likely to be fully protonated; in this state, X=NH3+.Cl– is an even better electron withdrawing group. The effect is also much stronger if X = equatorial. So one can predict here that if the alternate stereoisomer with X = axial were to be synthesised, it would hydrolyse more quickly. Other groups (X=F, CN etc) would probably show similar behaviour.‡
‡ I have added two further entries, X=CF3 and X=SF5 in the table above, showing the latter to be more effective at inhibiting hydrolysis.
Acknowledgments
This post has been cross-posted in PDF format at Authorea.
Author
References
- C.O. Ndubaku, J.J. Crawford, J. Drobnick, I. Aliagas, D. Campbell, P. Dong, L.M. Dornan, S. Duron, J. Epler, L. Gazzard, C.E. Heise, K.P. Hoeflich, D. Jakubiak, H. La, W. Lee, B. Lin, J.P. Lyssikatos, J. Maksimoska, R. Marmorstein, L.J. Murray, T. O’Brien, A. Oh, S. Ramaswamy, W. Wang, X. Zhao, Y. Zhong, E. Blackwood, and J. Rudolph, "Design of Selective PAK1 Inhibitor G-5555: Improving Properties by Employing an Unorthodox Low-p <i>K</i> <sub>a</sub> Polar Moiety", ACS Medicinal Chemistry Letters, vol. 6, pp. 1241-1246, 2015. https://doi.org/10.1021/acsmedchemlett.5b00398
- P. Camilleri, D. Munro, K. Weaver, D.J. Williams, H.S. Rzepa, and A.M.Z. Slawin, "Isoxazolinyldioxepins. Part 1. Structure–reactivity studies of the hydrolysis of oxazolinyldioxepin derivatives", J. Chem. Soc., Perkin Trans. 2, pp. 1265-1269, 1989. https://doi.org/10.1039/p29890001265
- P. Camilleri, D. Munro, K. Weaver, D.J. Williams, H.S. Rzepa, and A.M.Z. Slawin, "Isoxazolinyldioxepins. Part 1. Structure–reactivity studies of the hydrolysis of oxazolinyldioxepin derivatives", J. Chem. Soc., Perkin Trans. 2, pp. 1929-1933, 1989. https://doi.org/10.1039/p29890001929
- Beckmann, C.., Jones, P.G.., and Kirby, A.J.., "CCDC 209989: Experimental Crystal Structure Determination", 2003. https://doi.org/10.5517/cc71hvl
- C. Beckmann, P.G. Jones, and A.J. Kirby, "<i>N,N,N</i>′,<i>N</i>′-Tetramethylstreptamine 2,4,6-orthoformate hydrochloride", Acta Crystallographica Section E Structure Reports Online, vol. 59, pp. o566-o568, 2003. https://doi.org/10.1107/s1600536803006287
- H.S. Rzepa, "C 6 H 14 O 5", 2015. https://doi.org/10.14469/ch/191581
- H.S. Rzepa, "Gaussian Job Archive for C6H14O5", 2015. https://doi.org/10.6084/m9.figshare.1599751
- H.S. Rzepa, "C 6 H 15 N 1 O 5", 2015. https://doi.org/10.14469/ch/191582
- H.S. Rzepa, "C6H15NO5", 2015. https://doi.org/10.14469/ch/191586
- H.S. Rzepa, "C 6 H 16 Cl 1 N 1 O 5", 2015. https://doi.org/10.14469/ch/191584
- H.S. Rzepa, "C6H16ClNO5", 2015. https://doi.org/10.14469/ch/191588
- H.S. Rzepa, "C 6 H 16 Cl 1 N 1 O 5", 2015. https://doi.org/10.14469/ch/191590
- H.S. Rzepa, "Gaussian Job Archive for C6H16ClNO5", 2015. https://doi.org/10.6084/m9.figshare.1601891
- H.S. Rzepa, "C 7 H 13 F 3 O 5", 2015. https://doi.org/10.14469/ch/191592
- H.S. Rzepa, "Gaussian Job Archive for C7H13F3O5", 2015. https://doi.org/10.6084/m9.figshare.1603088
- H.S. Rzepa, "C 6 H 13 F 5 O 5 S 1", 2015. https://doi.org/10.14469/ch/191595
- H.S. Rzepa, "Gaussian Job Archive for C6H13F5O5S", 2015. https://doi.org/10.6084/m9.figshare.1603420
Tags: Cambridge, Derek Lowe, HTML, HTML element, medchem, medical chemistry
This is a plot of dipole moment vs IRC for the system X = NH3+.Cl- which I had not spotted when I wrote the post as unusually interesting.
Note that in the immediate transition state region (IRC = 0.0), the dipole moment changes dramatically. This is due to the two proton transfers which occur, and which have this large effect on the dipole moment. Which goes to show that without at least some correction for solvation, the potential energy surface of such reactions computed in the gas phase may be quite misleading.
Why even invoke a concerted hydrolysis mechanism? All of the textbooks show this going through a multi-step process that starts with protonation followed by oxonium formation.