The knowledge that substituents on a benzene ring direct an electrophile engaged in a ring substitution reaction according to whether they withdraw or donate electrons is very old.[1] Introductory organic chemistry tells us that electron donating substituents promote the ortho and para positions over the meta. Here I try to recover some of this information by searching crystal structures.
I conducted the following search:
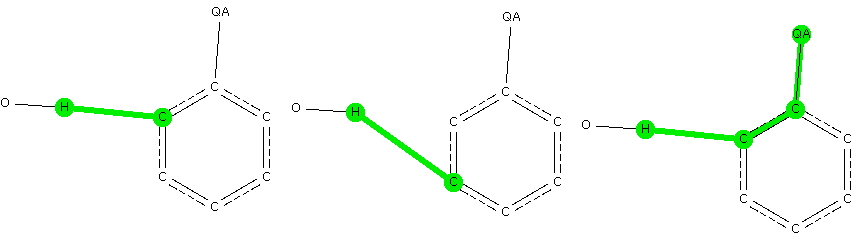
- Any electron donating group as a ring substituent, defined by any of the elements N, O, F, S, Cl, Br.
- A distance from the H of an OH fragment (as a hydrogen bonder to the aryl ring) to the ortho position relative to the electron donating group.
- A similar distance to the meta position.
- The |torsion angle| between the aryl plane and the C…H axis to be constrained to 90° ± 20.
- Restricting the H…C contact distance to the van der Waals sum of the radii -0.3Å (to capture only the stronger interactions)
- The usual crystallographic requirements of R < 0.1, no disorder, no errors and normalised H positions.
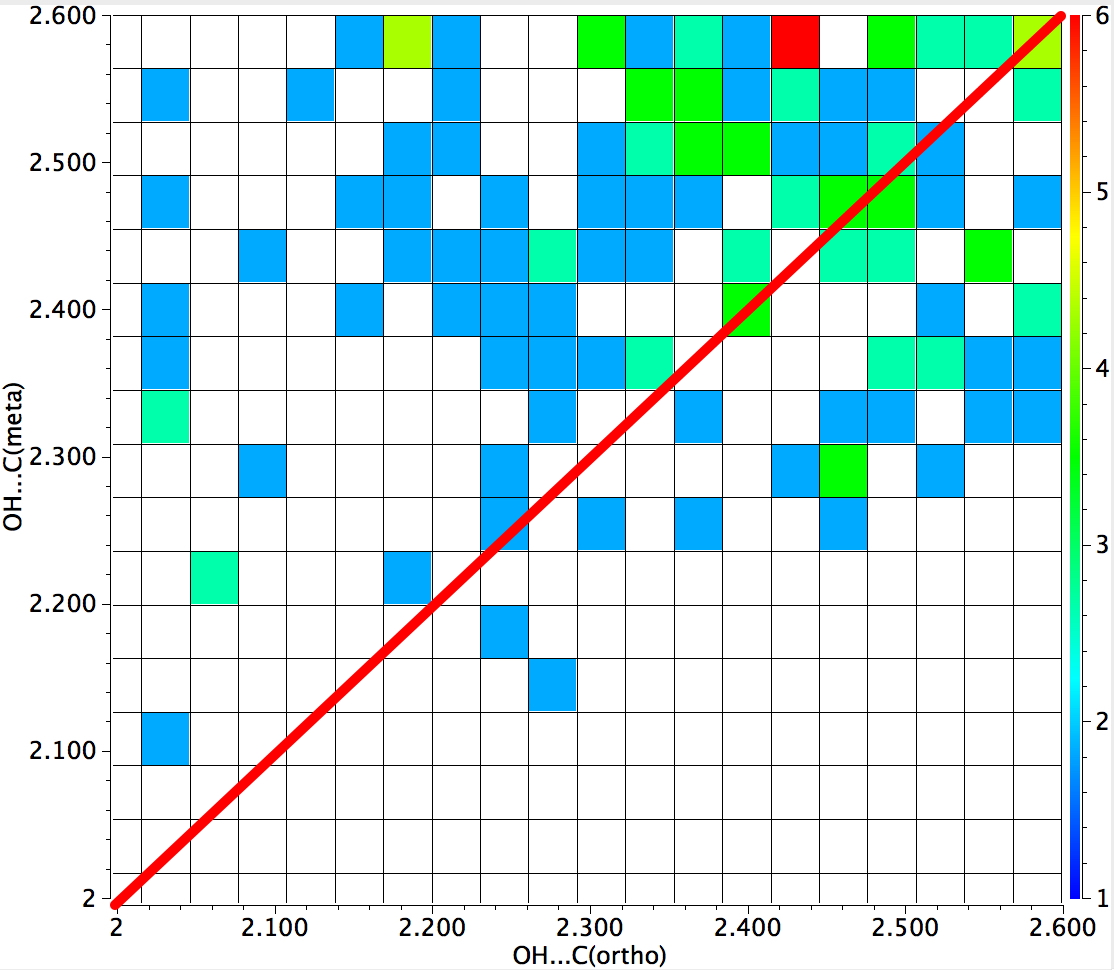
The result of such a search is seen below. The red line indicates those hits where the distance from the H to the ortho and meta positions is equal. In the top left triangle, the distance to ortho is shorter than to meta (and the converse in the bottom right triangle). You can see that both the red hot-spot and indeed the majority of the structures conform to ortho direction (of π-facial ) hydrogen bonding.
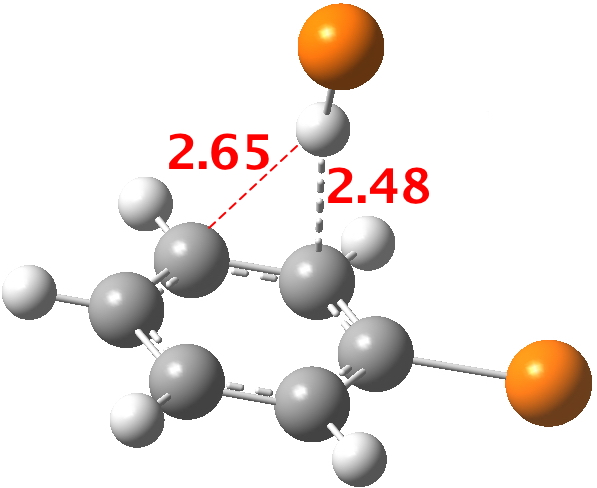
 Here is a little calculation, optimising the position that HBr adopts with respect to bromobenzene. You can see that the distance discrimination towards ortho is ~ 0.17Å, a very similar value to the "hot-spot" in the diagram above.
Here is a little calculation, optimising the position that HBr adopts with respect to bromobenzene. You can see that the distance discrimination towards ortho is ~ 0.17Å, a very similar value to the "hot-spot" in the diagram above.
This little search of course has hardly scratched the surface of what could be done. Changing eg the OH acceptor to other electronegative groups. Restricting the wide span of N, O, F, S, Cl, Br. Probing rings bearing two substituents. What of the minority of points in the bottom right triangle; are they true exceptions or does each have extenuating circumstances? Why do many points actually lie on the diagonal? Can one correlate the distances with the substituent? Is there a difference between intra and intermolecular H-bonds? What of electron withdrawing groups?
The above search took perhaps 20 minutes to define and optimise, and it gives a good statistical overview of this age-old effect. It is something every new student of organic chemistry can try for themselves! If you run an introductory course in organic aromatic chemistry, or indeed a laboratory, try to see what your students come up with![2]
Acknowledgments
This post has been cross-posted in PDF format at Authorea.
Author
References
- H.E. Armstrong, "XXVIII.—An explanation of the laws which govern substitution in the case of benzenoid compounds", J. Chem. Soc., Trans., vol. 51, pp. 258-268, 1887. https://doi.org/10.1039/ct8875100258
- H.S. Rzepa, "Discovering More Chemical Concepts from 3D Chemical Information Searches of Crystal Structure Databases", Journal of Chemical Education, vol. 93, pp. 550-554, 2015. https://doi.org/10.1021/acs.jchemed.5b00346
Tags: above search, Aromatic compounds, aromaticity, Birch reduction, Chemistry, electron donating, Electrophile, Electrophilic aromatic substitution, Ether, Functional groups, little search, Organic chemistry, Physical organic chemistry, Substitution reactions
I felt I had to follow up that quick exploration of EDG (electron donating groups) with the counterbalance of EWG (electron withdrawing groups).