We are approaching 1 million recorded crystal structures (actually, around 716,000 in the CCDC and just over 300,00 in COD). One delight with having this wealth of information is the simple little explorations that can take just a minute or so to do. This one was sparked by my helping a colleague update a set of interactive lecture demos dealing with stereochemistry. Three of the examples included molecules where chirality originates in stereogenic centres with just three attached groups. An example might be a sulfoxide, for which the priority rule is to assign the lone pair present with atomic number zero. The issue then arises as to whether this centre is configurationally stable, i.e. does it invert in an umbrella motion slowly or quickly. My initial intention was to see if crystal structures could cast any light at all on this aspect.
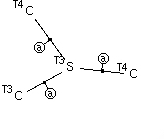
Central atom has three bonded atoms as C, of which either all three must themselves have four attached atoms, or one can have just three attached atoms as shown above, along with acyclic character for the three bonds attached to the central atom, R ≤ 0.1, not disordered and no errors.
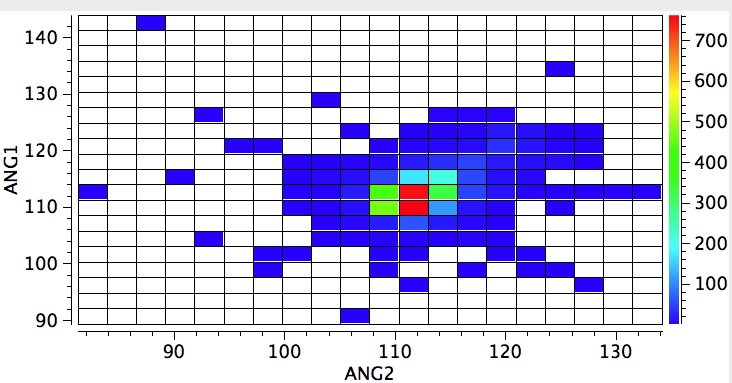
Using the search definition above for R3N one gets the result below. It shows a hot spot for an angle subtended at the nitrogen of ~111°, indicating a pyramidal nitrogen. But how easily is that perturbed? (which is almost like asking how easily can it invert its configuration?).
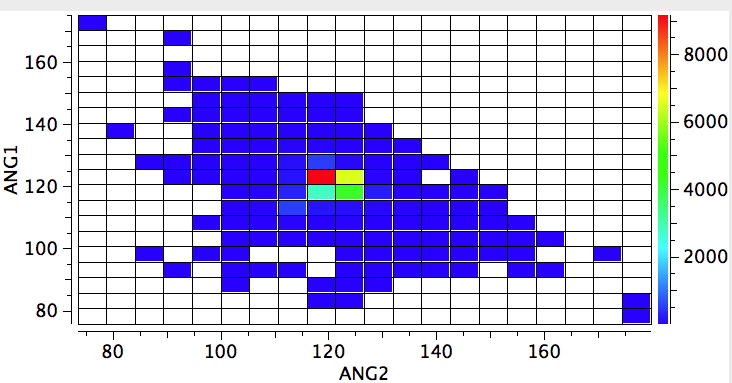
A perturbation can be applied by changing just one of the attached carbons as having three attached atoms of its own (sp2 hybridised). The response is that the hot spot moves to 120° (below). Of course now this includes compounds such as amides and the like. But we have learnt that it takes just one such attached sp2 hybridised carbon to planarize an adjacent nitrogen.
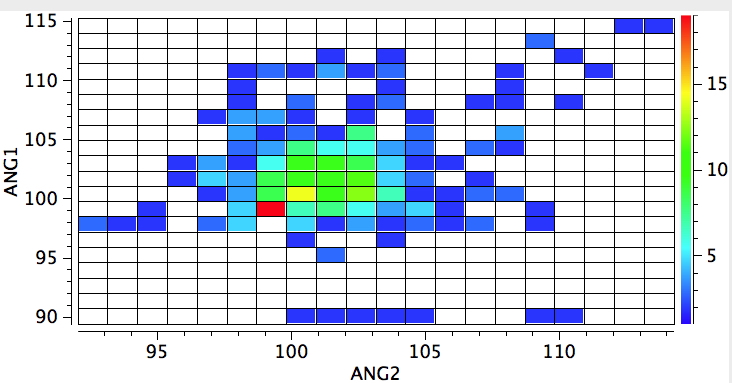
The control experiment will now be to apply the same test to a P. The hot spot moves from ~99° (P with three sp3 carbons attached) to ~103° (P with two sp3 and one sp2). This reminds us that the overlap and energy-match between a p-orbital on carbon to an adjacent p-orbital on nitrogen is good, whereas the same overlap/energy match to a p-orbital on P is significantly less so.
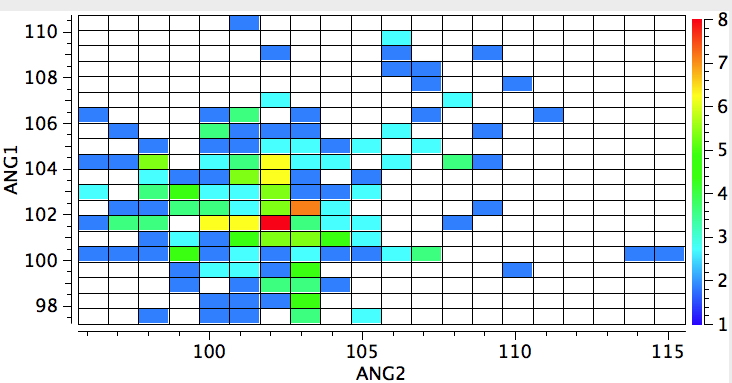
One gets the same result when the central atom is S; the hotspot moves from ~102° to ~105°. Unfortunately, not enough compounds are known for a tri-substituted oxygen compounds to see how this element responds.
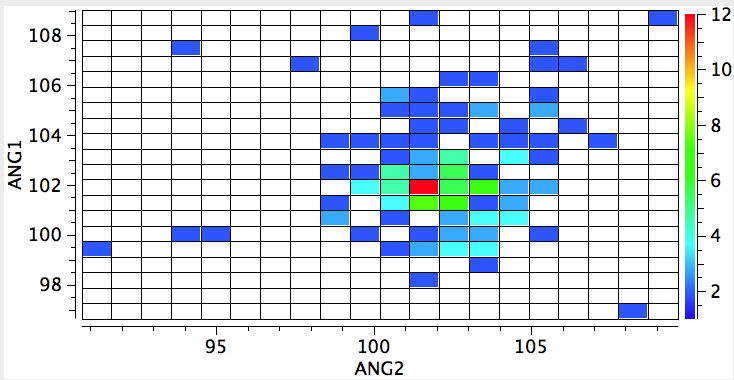
My point in illustrating these statistics is to show how much text-book chemistry can be recovered simply by a few quick explorations of crystal structures. One could even argue that much introductory chemistry could be taught by reference to the statistics of such structures.
Acknowledgments
This post has been cross-posted in PDF format at Authorea.