I am attending a conference. Plenaries at such events can sometimes provide interesting pointers on things to come (and sometimes they simply point to things past). At WATOC2014 in Santiago Chile, the first plenary was by Paul Ayers with the impressive title “Concepts for organising chemical knowledge” which certainly sounds as if it is pointing forward!
WATOC stands for World Association of Theoretical and cOmputational Chemists (yes, the acronym does not expand) and so the organising concepts of course relate to quantum mechanics. Ayer’s talk however was more about chemical philosophy, and how we may continue to organise chemical rules according to sound quantum mechanical principles. He bases these principles on observables, rather than just mathematical formulations. The start point is electron distributions (I have noted elsewhere that these are indeed measurable observables, although the crystallography required to measure bond-based electron density is only rarely carried out) in the form of conceptual DFT. He strives to find examples that cannot be explained by e.g. the older ideas of molecular orbitals (as obtained from solution of a single reference determinant) and in particular to avoid the modern syndrome that as we improve our ability to achieve numerical accuracy, we tend to lose our insight into the phenomenon being studied. One example he gives is that of alkynes such as FC≡CF. If one removes a π-electron from the molecule, this should result in a decrease in the electron density distribution? Wrong. It actually increases the σ-electron density in the nodal planes of the lost π-electron. Ayer’s point is that Conceptual DFT provides a simple framework for explaining such observations, unlike MO theory.
But the most interesting moment came with discussion of Bader’s QTAIM methods (often used in this blog), as used by Ayers to provide a formulation of an electronic stress tensor approach to molecules. Expressed as Ehrenfest Forces and the corresponding Hessian, these provides a handle on the forces on bonds, or in simpler language, telling us “how bonds might move“. Think of it as the electronic equivalent of how nuclei move in molecules (aka vibrations). The reason my ears pricked up is that I have often discussed how organic chemists tend to conceptualize “how bonds might move” by using curly arrow pushing. This concept, dating back to Robinson’s original example is often dismissed by theoreticians (and it has to be said by some organic chemists) as having little sound theoretical foundation; it is pure symbolism, and not to be over-interpreted. So perhaps electronic stress tensors based on Ehrenfest Forces might indeed tell us whether the hugely successful curly arrow pushing formalism (itself based on Lewis’ ideas of the shared electron bond formulated in 1916) might yet receive the theoretical makeover that it badly needs.
But before I end, one more example given by Ayers; keto-enol tautomerism in say propanone. Here a C-H bond breaks and an OH bond forms. The Ehrenfest Hessian indeed tells is that this is indeed “how the bonds want to move“. At the point the “audience” (in the form of a question) injected that sense of excitement that a conference occasionally provides. According to MO theory (!), the [1,3] sigmatropic shift of a hydrogen atom is a forbidden pericyclic process! How might the conceptual DFT electronic stress tensor approach tell us this? I have to confess I am not sure I understood Ayers’ answer at this point. Perhaps more work is needed before MOs are discarded entirely.
Oh, one more comment. Conference talks, presented in the form of slides which might linger on the projected screen for only a few seconds, rarely allow the audience to take much more than cursory notes. In particular, without some more research, I cannot here cite any of the references to Ayer’s recent work, since I did not have time to make a note of them.
Postscript: An article by Paul Geerlings, Paul Ayers et al on the topic of the Woodward-Hoffmann rules has in fact been published[1].
Postscript2: I have computed the density difference isosurface for FCCF as noted above. It is shown as a thumbnail below (blue is decreased density on forming the cation, red is the increased density). Click on this to rotate.
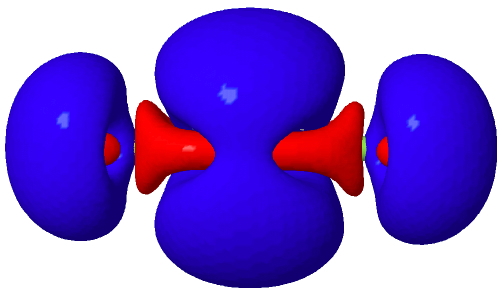
Click for 3D
Author
References
- P. Geerlings, P.W. Ayers, A. Toro-Labbé, P.K. Chattaraj, and F. De Proft, "The Woodward–Hoffmann Rules Reinterpreted by Conceptual Density Functional Theory", Accounts of Chemical Research, vol. 45, pp. 683-695, 2012. https://doi.org/10.1021/ar200192t
Tags: chemical knowledge, chemical philosophy, chemical rules, Chile, Paul Ayers, Paul Geerlings, Santiago, World Association of Theoretical and cOmputational Chemists