By about C17H36, the geometry of “cold-isolated” unbranched saturated alkenes is supposed not to contain any fully anti-periplanar conformations. [1] Indeed, a (co-crystal) of C16H34 shows it to have two-gauche bends.[2]. Surprisingly, the longest linear alkane I was able to find a crystal structure for, C28H58 appears to be fully extended[3],[4] (an early report of a low quality structure for C36H74[5] also appears to show it as linear).‡ Here I explore how standard DFT theories cope with these structures.
I start with noting the use of a TZVP basis set. In a recent article[6] we noted that the basis-set-superposition-errors for this basis were about a quarter of that for the standard Pople-type 6-311G(d,p) basis that I tend to use for modelling in this blog. This matters, since the relative energy of a folded-conformation vs an extended linear one might depend on the quality of the basis set and its inherent BSSE. The DFT method is the classical B3LYP. I also modelled C58H118 as the hydrocarbon as being well beyond the region anticipated above for folding of the chain to have started (no, there is no crystal structure). The geometries of linear and bent forms are shown below.

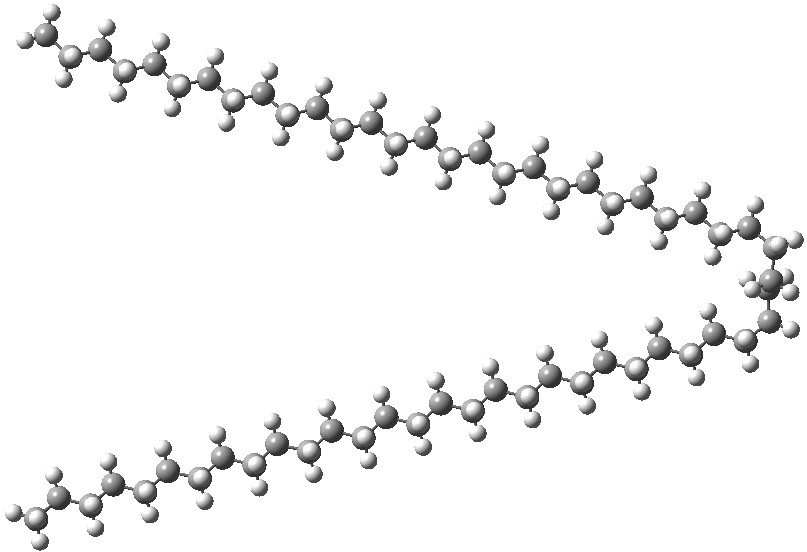
The relative free energy of the V-shaped bent form[7] emerges as 3.5 kcal/mol higher than the linear form[8]. Now, to add a Grimme-D3 dispersion correction to the energies. The V-shape of the bent form now adopts the hairpin mode,[9] and its energy is now 2.5 kcal/mol lower than the linear form.[10]
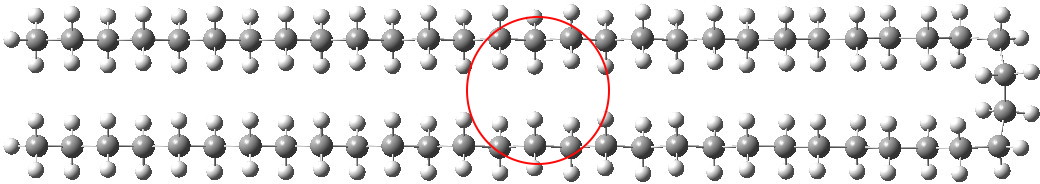
Note in the above the very slight strange oscillation (kink) that appears about 11 atoms away from the hairpin bend. I repeated this with the wB97XD DFT procedure (in which dispersion is implicit) and found the same result.
As triple-ζ basis quality modelling of molecules with >100 atoms becomes increasingly common, it is worth repeating yet again that the model should always contain dispersion (and solvent if appropriate) corrections as default. Indeed, it is probably also worth re-investigating much early modelling (by this I mean modelling done ten or more years ago) to see if such corrections significantly influence the conclusions.[6]
‡The searches cannot be carried out according to the formula CnH2n+2, but must be done individually for the value of n. I gave up at C50.
Author
References
- N.O.B. Lüttschwager, T.N. Wassermann, R.A. Mata, and M.A. Suhm, "The Last Globally Stable Extended Alkane", Angewandte Chemie International Edition, vol. 52, pp. 463-466, 2012. https://doi.org/10.1002/anie.201202894
- N. Cocherel, C. Poriel, J. Rault‐Berthelot, F. Barrière, N. Audebrand, A. Slawin, and L. Vignau, "New 3π‐2Spiro Ladder‐Type Phenylene Materials: Synthesis, Physicochemical Properties and Applications in OLEDs", Chemistry – A European Journal, vol. 14, pp. 11328-11342, 2008. https://doi.org/10.1002/chem.200801428
- S.C. Nyburg, and A.R. Gerson, "Crystallography of the even <i>n</i>-alkanes: structure of C<sub>20</sub>H<sub>42</sub>", Acta Crystallographica Section B Structural Science, vol. 48, pp. 103-106, 1992. https://doi.org/10.1107/s0108768191011059
- R. Boistelle, B. Simon, and G. Pèpe, "Polytypic structures of n-C28H58 (octacosane) and n-C36H74 (hexatriacontane)", Acta Crystallographica Section B Structural Crystallography and Crystal Chemistry, vol. 32, pp. 1240-1243, 1976. https://doi.org/10.1107/s0567740876005025
- H.M.M. Shearer, and V. Vand, "The crystal structure of the monoclinic form of n-hexatriacontant", Acta Crystallographica, vol. 9, pp. 379-384, 1956. https://doi.org/10.1107/s0365110x5600111x
- A. Armstrong, R.A. Boto, P. Dingwall, J. Contreras-García, M.J. Harvey, N.J. Mason, and H.S. Rzepa, "The Houk–List transition states for organocatalytic mechanisms revisited", Chem. Sci., vol. 5, pp. 2057-2071, 2014. https://doi.org/10.1039/c3sc53416b
- H.S. Rzepa, "Gaussian Job Archive for C58H118", 2014. https://doi.org/10.6084/m9.figshare.978501
- H.S. Rzepa, "Gaussian Job Archive for C58H118", 2014. https://doi.org/10.6084/m9.figshare.978502
- H.S. Rzepa, "Gaussian Job Archive for C58H118", 2014. https://doi.org/10.6084/m9.figshare.978832
- H.S. Rzepa, "Gaussian Job Archive for C58H118", 2014. https://doi.org/10.6084/m9.figshare.978833
Tags: dispersion, energy, relative energy, relative free energy