Text books will announce that during aromatic electrophilic substitution, aromaticity is lost by the formation of a Wheland intermediate (and regained by eliminating a proton). Is that entirely true?
I will start by considering the simplest of all such intermediates[1],[2] the NMR of which was first reported by Olah and which is shown below (red). The values in black are from a ωB97XD/6-311G(d,p) calculation (chloroform)[3]. The discrepancy might be due to the difference in solvent (HF-SbF5/SO2ClF-SO2F2) and my failure to include a counter-ion in the calculation.‡ Actually, this result is only obtained at -134°C; warming to -70°C increases the rate of [1,2] ring shifts and only one “aromatic” peak at 8.09 ppm is observed.
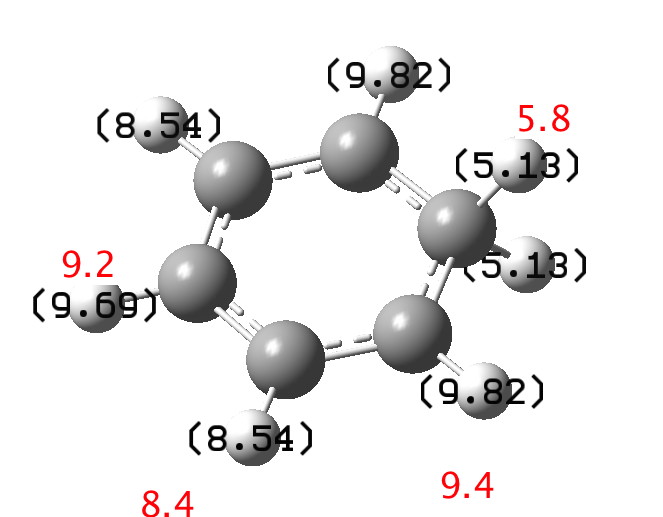
Click for 3D to see vibrational modes
The computed MOs are shown below. MOs 21 and 15 are pretty much identical with those for benzene itself (the former because of symmetry), so relatively little disruption of the conjugation there. An NBO analysis shows that each C-H bond from the sp3 centre acts as a donor to the π-system (E2 = 8.5 kcal/mol), which we know in another guise as hyperconjugation or σ-conjugation. In this sense, the two C-H bonds are acting as surrogates for a real p-AO on that carbon, and the E2 value is certainly not insignificant. Certainly they are nowhere near as good a donor as one real p-AO, but nonetheless, the two of them together are good enough to result in retention of a significant measure of cyclic π-conjugation, and hence probably aromaticity. The C-H bonds are weakened as a result of their electron donation, which reduces their normal mode wavenumber down by about 200 cm-1. The so-called Kekule-mode, which is depressed in benzene itself to about 1300 cm-1 because of the effect termed π-distortivity[4], is actually increased to 1442 cm -1 in the Wheland intermediate.[5] This mode is normally elevated by any weakening of the distortive π-system (in my previous post, I noted such an elevation induced by the quintet excited state of benzene) and so we many presume the same effect operates in the Wheland intermediate.
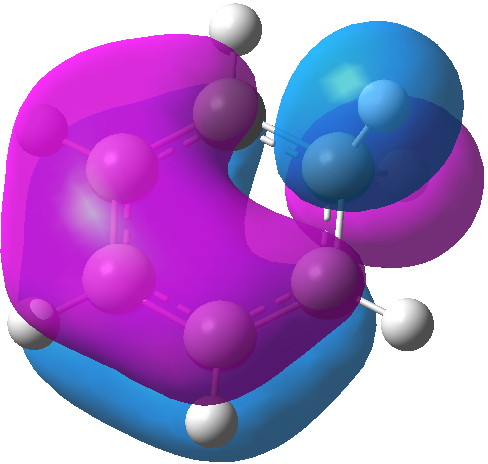 MO 20 Click for 3D |
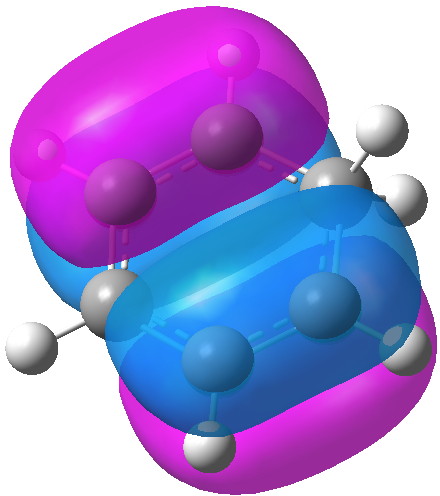 MO 21 Click for 3D |
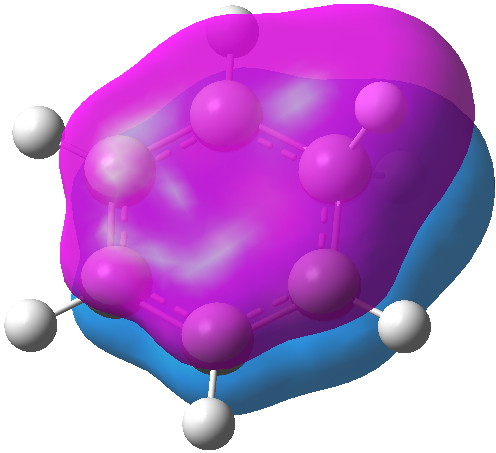 MO 15 Click for 3D |
|
To get a measure of any aromaticity deriving from a ring current, I computed the NICS(1) value from the ring centroid. This latter was itself obtained from the coordinates of the ring-critical-point determined by a QTAIM analysis, and computed at that point and 1, 2 and 3Å above it. The NICS values are respectively -0.4, -6.1, -3.6 and -1.6 ppm. The maximum is indeed the NICS(1) point, and at that point, the value indicates modest, but real π-aromaticity.
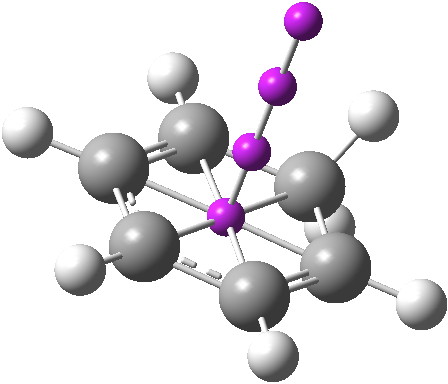
So does forming a Wheland intermediate disrupt all aromaticity? The answer is a clear no! What might be interesting to compute (I have not tried doing so here) is an actual energy for the stabilisation resulting from the weak aromaticity present as a % of that present in benzene, so that the text-books can be amended.
‡ It might also be that the exchange in the measured NMR spectrum was not entirely suppressed, and so some residual averaging of the peak positions remains.
Author
References
- G.A. Olah, R.H. Schlosberg, D.P. Kelly, and G.D. Mateescu, "Stable carbonium ions. IC. Benzenonium ion (C6H7+) and its degenerate rearrangement", Journal of the American Chemical Society, vol. 92, pp. 2546-2548, 1970. https://doi.org/10.1021/ja00711a057
- G.A. Olah, R.H. Schlosberg, R.D. Porter, Y.K. Mo, D.P. Kelly, and G.D. Mateescu, "Stable carbocations. CXXIV. Benzenium ion and monoalkylbenzenium ions", Journal of the American Chemical Society, vol. 94, pp. 2034-2043, 1972. https://doi.org/10.1021/ja00761a041
- S. Shaik, A. Shurki, D. Danovich, and P.C. Hiberty, "A Different Story of π-DelocalizationThe Distortivity of π-Electrons and Its Chemical Manifestations", Chemical Reviews, vol. 101, pp. 1501-1540, 2001. https://doi.org/10.1021/cr990363l
- H.S. Rzepa, "Gaussian Job Archive for C6H7(1+)", 2013. https://doi.org/10.6084/m9.figshare.870473
Tags: actual energy
Mulliken pointed out long ago that CH2 groups in C6H7(+) hyperconjugate (cf., Wu, Pure Appl. Chem, 2013, 85, 921). However, CHX groups in C6H6X+ species (where X is electronegative) do not and are not aromatic (cf., ACIE, 2011, 50, 6089)
NICSpizz (OL, 2006, 8, 863) is much more refined than isotropic NICS.
Thanks for the background Paul. Yes, more to come on the better analysis of the aromaticity, and also the vibrations.
As for X=electronegative, yes they would release electrons more poorly. Conversely, electropositive X (Si?) might enhance the effect (Li?). In fact you have calculated so much Li in the past, I am sure that C6H5Li2(+) must be in your archive? Is the Li(+) bound to the centre of the benzene ring?
In the post, I mentioned that the Wheland intermediate Kekule mode was elevated compared that of benzene. To be more accurate, one needs to remove the mass weighting from these frequencies. One can do this with a little trick in fact, which is to reduce the mass of the hydrogens to almost zero.
Now, the Kekule mode of benzene comes out as 1317, and that of its Wheland intermediate 1504. So the exaltation of this mode due to the weakening of the π-system is still real.
Here is an odd result. I calculated the dilithio system and did the reduced mass trick (using the kekule mode as a scale of aromaticity). I got 1278 cm-1 for this mode, even more depressed than benzene itself.
But a system well worth reporting on later. I note for the time being that all six C-C bonds in this system are very close to 1.40Å, so it is certainly aromatic.
I try effect of hyperconjugation and replace CH2 for CMe2. There was no effect on anizotropy: details
Another variation is to replace the H-C-H group with a H-B-H, which results in greater electron release into the aromatic ring. The computed MOs resemble those of benzene itself far more closely than the benzene Wheland, and the Kekule mode is 1377 cm-1, only modestly elevated compared to benzene itself. One might speculate whether this (neutral) analogue of the Wheland intermediate is capable of being synthesised.
Re: replace CH2 for CMe2. I do recollect a crystal structure where CH2 is replaced by C(SiR3)2. See doi: 10.1039/b513203g. The counterion is BAr4(-) and the structure is shown below. Note that the bond to the 4-substituted ring carbon is only slightly longer than the others, and highly typical of an aromatic bond. So we may presume that C-Si is also very electron releasing.
Combining these themes, one might further speculate that B(SiR3)2 would be an even better group at aromatising the “wheland intermediate”. Anyone out there care to make it? I guess the spoiler might be those [1,2] rearrangements, which will rearrange the molecules away from these optimal cases.
Based on NICS, Wheland intermediates certainly do seem to suffer from a substantial disruption of aromaticity. A comparison of the computed dissected NICS(0)πzz for benzene (–35.9 ppm), protonated benzene (–18.0 ppm), and the brominated Wheland intermediate, C6H6Br(+), (+3.1 ppm) are illustrative. C6H5Li2(+) would be an interesting one to look into too.
Dear all,
in terms of current topology the Wheland cation, much to my surprise, looks like benzene. With a nice strong diatropic flow around the whole molecule and in the inside a substantial paratropic curl.
Also as foreseen by Mulliken, cyclopentadiene is hyperconjugatively like arenium ions (also with regard to methylene substitution). See Hyperconjugative -Aromaticity: How to Make Cyclopentadiene Aromatic. Laszlo Nyulaszi. and Paul von Rague Schleyer. J. Am.Chem.Soc., 1999, 121, 6872-6875. doi: 10.1021/ja983113f
Thanks for explanations and references Paul!
The usage of the “ring critical point” comes imho a bit out of the blue, as there is no direct connection between the electron density topology and the the current topolgy _in general_ known as yet. I prefer in such cases the use of the central current stagnation point [r_0 for which j(r_0) = 0].
Finding a general relation between the two would be high interest!
Re: “I prefer in such cases the use of the central current stagnation point [r_0 for which j(r_0) = 0].”
Yes, that sounds a more pertinent point. But one does need specialised programs to compute this; QTAIM is rather more generally available.
Perhaps you can suggest an easily available program for computing current stagnation points?
GIMIC interfaces with Turbomole and ACESII.
Get it like that:
git clone https://github.com/bakerjonas/gimic.git
cd gimic
./setup
cd build
make
sudo make install
You can calculate vector fields like that one shown in the picture above. Searching the stagnation points “by hand” using “nested intervals” works pretty well for me1. Advanced computer users can also write some Newton-Raphson optimizer script for the inverse modulus of j (only a few lines in python f. i.).
Paul notes his 1999 investigation of cyclopentadiene, in which the two methylene hydrogens are replaced by SnR3 groups. To link into the above, I have done the same, but replacing the hydrogens with two Lithium atoms (doi: 10.6084/m9.figshare.872568 ). The resulting structure has all the C-C bond lengths almost equal, but a more interesting partitioning of the four electrons nominally associated with the two “C-Li” bonds. Two enter the ring to achieve 6-π-electron aromaticity for the ring, the other two form an in-plane σ-carbene. This species might be a very strong base as a result!
The three highest occupied π-orbitals again strongly resemble those of benzene. The ELF analysis (below) shows the ring to contain five ~3-electron basins and the carbene lone pair (label 15, 2.6 electrons).
There is also a handfull of other programms capable of calculating currents. Keith Todd’s AIMALL being one of them.
This and othe issues concerning molecular currents are discussed in our facebook group called “Magnetically Induced Electronic Currents in Molecules”. Anyone interested is cordially invited to join the group. Among our members are Jeppe Olsen, Trygve Helgaker, Kenneth Ruud, Jochen Autschbach and many more.
Here is another analogue, with BH2 replacing CH2 in cyclopentadiene, forming an anion. It too has some measure of apparent aromaticity (but less than some of the other systems discussed above).
How to count these hyperconjuagted electrons? 2? 4?
What trends in terms of aromaticity are to be expected in the series cyclpropene, cp, cycloheptatriene, … ?
Re: “What trends”. Well, nominally, hyperconjugation should be suppressed in cyclopropene (4n anti-aromaticity), encouraged in cyclopentadiene (see above, and Paul’s comment) and suppressed in cycloheptatriene. But the latter is bound to be non-planar, so it is not a fair progression.
Certainly the little tricks played above (BH2 for CH2, or CLi2 for CH2) should not succeed for cyclopropene.
Henry, We’ve explored this, too. E.g., the aromaticityl of 3,3-difluorocycloppropene, where the CF2 group acts like a HC+ moiety.
See A Study of Aromatic Three Membered Rings HJ Wang, P. v. R. Schleyer, J. I. Wu, Y. Wang, H. J. Wang Int. J. Quant. Chem. 2011,111, 1031-1038
Hello all, here is another good reference where we discussed hyperconjugative aromaticity/antiaromaticity in cyclopropene, cyclopentadiene, cycloheptriene, and cyclononatetraene (I. Fernandez, J. I. Wu, P. v. R. Schleyer, Substituent Effects on “Hyperconjugative” Aromaticity and Antiaromaticity in Planar Cyclopolyenes, Org. Lett. 2013, 15, 2990-2993.) Within this (CH)nCX2 (n = 2, 4, 6, 8, X = H, SiH3, F) set, the saturated CH2 and C(SiH3)2 groups act as “pseudo” 2 pi electrons, CF2 groups do the opposite and induce an “empty” p orbital at the saturated C site. Check out the alternating trend we presented in the paper. It’s quite interesting!
Henry,
We’ve also published on C5H4Li2, which you examined above, along with its group 14 congeners:
Aromaticity in group 14 metalloles: Structural, energetic, and magnetic criteria
Author(s): Goldfuss, B; Schleyer, PV ORGANOMETALLICS 16 Pages: 1543-1552 DOI: 10.1021/om960994v 1997
I have quickly checked out the currents in cyclopropene and cp. cp has the a relatively strong paratropic current, but it does not overcome the diatropic one. cyclopentadiene sustains all in all just a weak ring current. cycloheptatriene is a bit more difficult to integrate, lets see…
Ralph
Please check out the currents in cyclopropane and planar cyclobutane, which have been discussed as sigma aromatics and anti-aromatics, respectively.
3,3-difluorocyclopropene and 5,5-disilacyclopentadiene should be appreciably more aromatic than their hydrocarbon parents, as Judy has pointed out.