The mechanism of ester hydrolysis is a staple of examination questions in organic chemistry. To get a good grade, one might have to reproduce something like the below. Here, I subject that answer to a reality check.

In this scheme, HA is a general acid, R=Me, and the net result is to break what is called the acyl-oxygen bond (red). The mechanism is actually incomplete, since the label PT designates a proton-transfer (the mechanism for which is left somewhat undefined). Additionally, a lot of charges come and go and five steps or so are involved. So a student might be tempted to “fast-track” the whole process. Below I show two such fast-tracks (I prefer to say simplifications):

In the blue mechanism, the role of HA is actually played by one water molecule, and a second water is assisting the PT step (a far more thorough analysis of the mechanism can be found in this reference[cite]10.1139/V09-011[/cite]). The reaction is bimolecular in ester and the HA (=water in this case). The third water would make it a termolecular reaction overall, but if the reaction takes place in water itself than [H2O] would be constant. It would correspond to what the text books call AAC2 since we consider one molecule as an acid HA. But, one could look at it differently and consider the second water as a nucleophile generated by concurrent deprotonation (by the first water). This would make it a BAC2 type. It turns out that if one makes the mechanism cyclic, the AAC2 and BAC2 annihilate each other in effect to create a single (peri)cyclic mechanism (which has no well known name, but might be referred to as the co-operative pathway). Such a mechanism can be extended using a third water molecule (magenta diagram); I will come to the reason for including that presently.
Why would one want to even consider such mechanisms? Because, if you look carefully, you will see no charges! Charge separation (= large dipole moment) takes energy. It is normally thought that this energy is more than compensated for by additional solvation (a process which is implicit rather than explicitly shown in text-book diagrams). But if you do not generate charge separation, you might not need that solvation energy. I will turn to quantum mechanics to try to decide what might be viable (I hesitate to use the term “going on”).
A ωB97XD/6-311G(d,p)/SCRF=water model (in which solvation is approximately included as a continuum model) calculation yields the following for the blue mechanism.
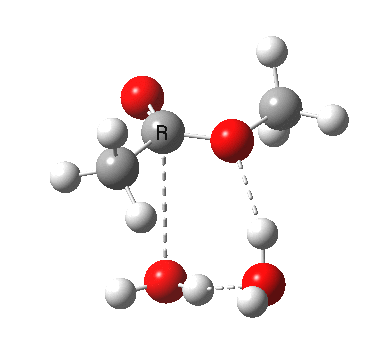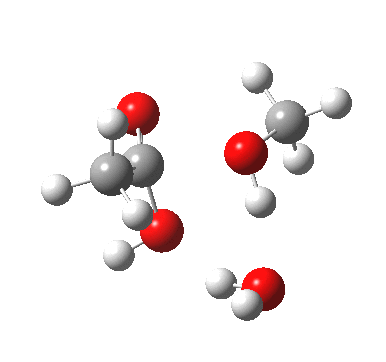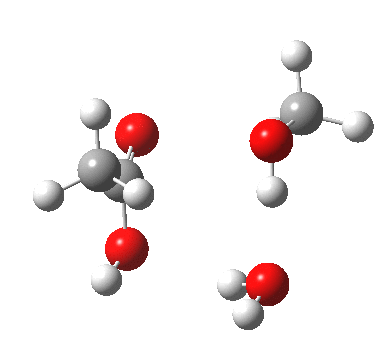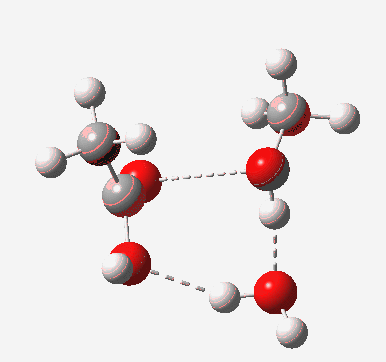 [cite]10.6084/m9.figshare.661351[/cite] [cite]10.6084/m9.figshare.661351[/cite] |
 |
- Points to note are that it is concerted, in other words the quantum mechanics tells us that all the bonds CAN make and break in a single concerted process within a single kinetic step.
- The mechanism has an uncanny resemblance to the nucleophilic aromatic substitution I reported a couple of posts ago! It resembles an Sn2 displacement at an sp2 centre. Such juxtaposition of these two mechanisms is also not found in text-books. Recollect that with such aromatic substitution, it was possible to get both cncerted and stepwise mechanisms, depending on the substituents. Perhaps the same might be possible here?
- However, the energy barrier for the process with the substituents shown above (~45 kcal/mol) is rather too high (the experimental value is estimated as >22 kcal/mol[cite]10.1139/V09-011[/cite]). There may be at least three reasons for this;
- (a) a better solvation model would be needed to lower the energy,
- (b) the angles subtended at the transferring protons are strained (they optimally should be linear) and
- (c) water is a very poor general acid (or base)!
But as an answer in an examination, would the blue mechanism actually be wrong? You will have to ask the instructor setting the question how they might respond to that, although these authors[cite]10.1139/V09-011[/cite] certainly conclude that such a concerted mechanism is the more “correct”, at least for hydrolysis in water without added acid or base.
Point (b) above can be addressed by adding another water molecule, as per the magenta mechanism so as to enlarge the ring and reduce the angular strain. But before I present the results, I need to “normalise” the system by ALSO adding one (solvating) water molecule to the blue route, as below, so that we can directly compare the energies of the blue and magenta pathways.
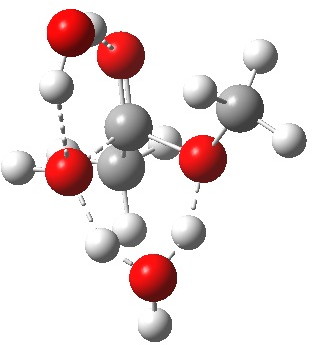
Click for 3D.
The result is a larger ring where the angular strain is clearly reduced. There is an entropic penalty for introducing that third water molecule, but despite this the free energy comes out 5.5 kcal/mol lower, and the activation barrier is also lower (~37 kcal/mol, still rather higher than experiment). It has been reported that incorporation of a 4th water molecule further improves matters[cite]10.1139/V09-011[/cite].
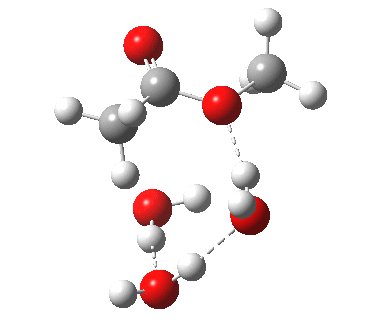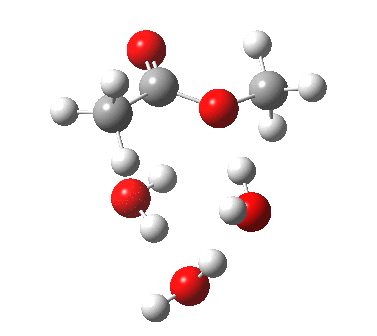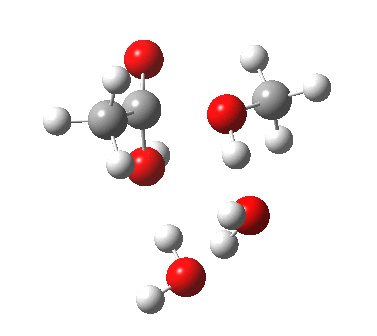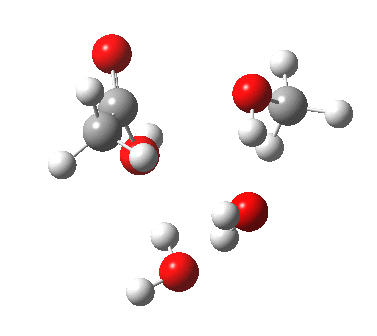 [cite]10.6084/m9.figshare.661789[/cite] [cite]10.6084/m9.figshare.661789[/cite] |
 |
We can also address both points (b) and (c) above by replacing HA=H2O by HA=guanidiniumH+ (green), a better general acid. This polar modification introduces the ability for the system to better sustain charge separations, and indeed the initial product is now an ion pair tetrahedral intermediate (methoxide anion and guanidinium cation) carrying a dipole moment of 14.5D, an increase over the value for the transition state with three waters, 9.7D. The barrier (~21 kcal/mol) has gone in the opposite direction, decreasing significantly compared to the water catalysed reaction. The tetrahedral intermediate sits in an energy well of ~4 kcal/mol.

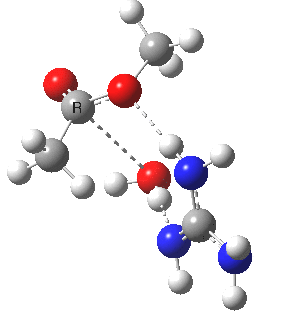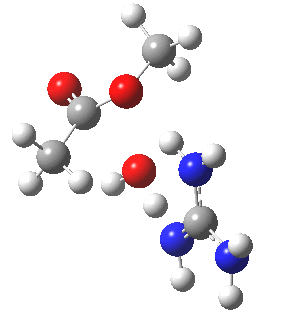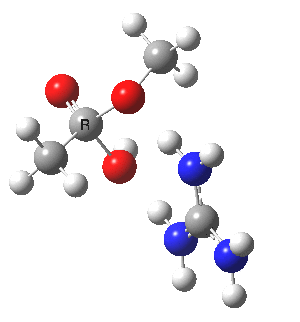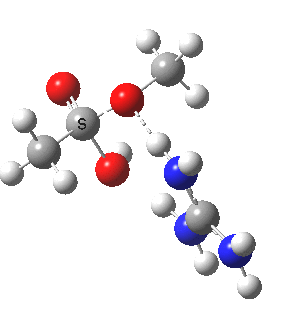 [cite]10.6084/m9.figshare.661791[/cite] [cite]10.6084/m9.figshare.661791[/cite] |
 |
A second transition state exiting the tetrahedral intermediate has a free energy barrier[cite]10.6084/m9.figshare.661799[/cite] about 2.5 kcal/ol lower than the one entering it.
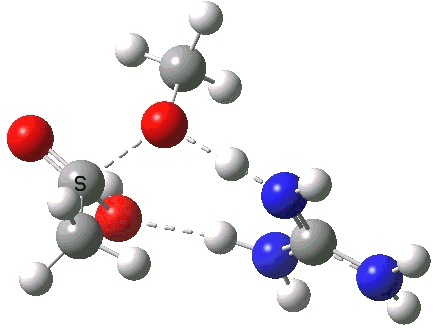
Click for 3D.
What might we have learnt? That ester hydrolysis using pure water could proceed through a cyclic and concerted transition state, involving three (or perhaps more) water molecules passing a proton baton along the chain, and in the process avoiding any large build up of charge separation. Replace two of these waters with say guanidine as a general acid/conjugate base capable of conjugatively stabilising charge-separated species and the mechanism changes to a stepwise reaction involving a dipolar tetrahedral intermediate sitting in a relatively shallow energy well.
Not possibly a picture that we might expect a student sitting an introductory examination in organic chemistry to reflect in its entirety, but also one that perhaps the text-books might start to hint at? Or: at some stage, armed merely with a “smart watch-cum-supercomputer”, a student taking such an exam might respond by performing the calculations described here as their submitted answer? Well, not for a year or two perhaps. But it has to be said that everything you see in this post was performed over less than two days of elapsed time, so these “reality checks” are not that time-consuming. Whether you choose to believe them or not of course is another matter.
Tags: ALSO, co-operative, energy, energy well, ester hydrolysis, free energy, Reaction Mechanism, shallow energy, solvation energy, Tutorial material
Just a few days ago I was thinking practically the same thing: with the overwhelming number of computational works showing that reactions involving a proton transfer in water solution are almost always assisted by two or more waters, may be it is time to start mentioning it in the textbooks?
By the way, is there a hidden intimidate at -5 in the IRC plot for the first reaction?
Water is indeed a remarkable (almost unique) proton transfer agent. It can simultaneously both donate and receive protons without requiring much of an energy barrier. Whilst it might seem that binding extra waters into a chain to do this would require reducing the entropy significantly, removing those waters from bulk water (where they are already held pretty tightly) is probably not such a problem. Regarding the hidden intermediate, the below is the gradient norm associated with the IRC. There is indeed a feature at IRC -3, but I do not think it qualifies as a proper “hidden intermediate” (the gradients never really approach being close to zero).


There is even less sign of a hidden intermediate for a chain of three water molecules:
In some cases, including acyl transfer reactions, we noticed that adding or removing extra water molecules may convert hidden intermediates into real minima, or vice versa. Similarly, in this case adding the third water removed that shoulder on the IRC. Regarding the entropic penalty for removing water molecules from bulk: to me, this looks more like just reorientation of the water molecules in the first solvating shell, rather than removal. There are water molecules that surround the ester molecule anyway, so it’s rather ‘differential solvation’ of the reagent vs the TS (I am stretching the concept a bit, by considering the assistance in transferring a proton as a special type of TS solvation). Something like umbrella sampling of a free energy along the C–O bond in a periodic water box with CPMD or with QM/MM with the first solvation shell in the QM part could be interesting, but quite challenging…
There is of course another mechanism for ester hydrolysis by pure water put forward by Gunaydin and Houk a few years ago: autoionization of water to form H3O+ and OH-, which then rapidly hydrolyze the ester through simultaneous acid and base catalysis. This readily explains the observed experimental barrier.
http://dx.doi.org/10.1021/ja8050525
Re “this looks more like just reorientation of the water molecules in the first solvating shell, rather than removal”. Participating in a proton transfer shell involves a much strong “hydrogen bond” then those found in a first solvating shell. So it is a matter of converting a “medium to weak” hydrogen bond into a significantly more rigid participation in a proton transfer ring.
I agree of course that a MD treatment is always the best option. Current computers probably need to speed up by a factor of 10-1000 to make this a viable routine option. Give it five years!
Re: “autoionization of water”. Ideally, one would want to set up a quantum mechanical model which allows whatever charge-transfer (or you could call it auto ionisation) to happen to whatever extent is energetically favourable. My model, which includes full geometric relaxation within a solvent continuum, may allow at least some extent of auto ionisation to occur (but admittedly limited by the lack of explicit solvent H-bonds etc). Such geometric relaxation was NOT automatically performed until relatively recently, largely because smoothed gradients within the continuum were not available.
So I would argue that geometric relaxation in a solvent model could certainly incorporate autoionisation. But as noted above, there are also dynamic/entropic effects associated with the first and outer solvent shells etc. Basically, a full complete model of reactions in which charge-separation is created/annihilated are a really tough nut to crack. But we are getting much closer to it, and as I note above, I would say more quantitative models which incorporate some of these refinements are probably no more than five years away from being routine. By routine, I mean something for which valuable insight can be obtained in about two days of computation.
‘Participating in a proton transfer shell involves a much strong “hydrogen bond” then those found in a first solvating shell. So it is a matter of converting a “medium to weak” hydrogen bond into a significantly more rigid participation in a proton transfer ring.’
Certainly! But this effect is (mostly) included in the free energy of activation calculated as a difference between the TS and the reagent cluster, so we don’t have to worry about it when relating the computational results to the reaction in bulk water.
‘So I would argue that geometric relaxation in a solvent model could certainly incorporate autoionisation.’
Indeed, looking at the IRC animations of water-assisted reactions shown above one may argue that these reactions are coupled to water autoionzation—and in fact it is the proton transfer between the waters what initiates the reaction.
Yes, I suppose one can regard autoionization in two ways; a trajectory on the dynamic pathway which results in a reaction (whilst trajectories that do not auto-ionise lead to different outcomes) and a feature on the intrinsic reaction coordinate that corresponds to the transient formation of an ion pair involving eg hydronium cation and hydroxyl anion.
[…] Chemistry with a twist « A sideways look at the mechanism of ester hydrolysis. […]
While I enjoy these reality checks by quantum mechanic calculations, maybe the results of some of those calculations need a reality check based on experimental data, as for example kinetic data. Based on this (didactic) article:
http://pubs.acs.org/doi/abs/10.1021/ed100345k
the pH profile of ester hydrolysis in either acidic or basic medium shows a correlation with H+ or OH- concentration, respectively. This makes a strong case for the importance of the traditional ionic pathways over the whole pH region, even in the neutral range. Participation of neutral mechanisms may be more important with other substrates, though. In any case, preparative ester hydrolysis is carried out under strongly acidic or basic conditions, so the mechanism is probably quite clearly ionic.
Autoionization of water may create highly reactive H+ and OH-, but at very low concentrations (defined by Kw). It might then be found that kinetic constants higher than the diffusion limit have to be assumed in order to get observable reaction rates – meaning that such pathways are usually kinetically excluded.
Also the earlier calculations on oxime formation, assuming neutral transition states and water chains are a bit disturbing in that respect, because classical kinetic studies (Jencks) make a strong case for ionic intermediates in those reactions, too.
http://pubs.acs.org/doi/abs/10.1021/ja01511a053
I think we should not abandon ionic mechanisms too quickly (in teaching, anyway; this was somewhat implied in the introduction of this blog post), just because they are more difficult to calculate with current quantum chemical methods.
The barriers reported above (~36 kcal/mol) for the pure water catalysed hydrolysis are significantly reduced to about 20 kcal/mol by introduction of a base/conjugate acid such as guanidine, which changes the mechanism from a concerted process to one proceeding through an (ionic) tetrahedral intermediate. So that certainly corresponds to experimental reality!
Some more observations on the autoionization of water. The Wikipedia article reminds of the timescale of this phenomenon, ~ 1ps, or the time it takes hydrogen bonds to reorganise. In fact, if a H3O(+) OH(-) pair is formed (very much a dynamic process) which happens to be separated by one or more neutral water molecules, a set of proton transfers will annihilate this ion pair in about 1ps. I set up a H3O(+).3H2O…OH(-).3H2O solvated ion pair in a ωB97XD/6-311+G(d,p)/SCRF=water potential, separated by one neutral water, and started to minimise the energy of the system. The dipole moment in this configuration was ~20D. As the optimisation proceeded, a cascade of proton transfers quickly reduced the dipole moment steadily, the last being the creation of eight H-bonded neutral waters.
So if such a transient H3O(+) OH(-) pair exists, it seems much more probable that it will annihilate by fast proton transfers than organise itself to participate in eg hydrolysing a ester. But of course if one adds additional species (such as the guanidine that I modelled above) we have an entirely different dynamic. I guess it really does boil down to what the dynamics of the system tell us about the probable trajectories, rather than just the nature of the potential energy surfaces.
[…] at room temperature) to ~11 kcal/mol (a very rapid reaction). A similar drop was noted for the hydrolysis of methyl ethanoate. But there is a more significant difference. With guanidine, “hidden intermediates” are […]
Apropos: http://pubs.acs.org/doi/abs/10.1021/jp311504d “Revisiting the Mechanism of Neutral Hydrolysis of Esters: Water Autoionization Mechanisms with Acid or Base Initiation Pathways”