Consider acetaldehyde (ethanal for progressive nomenclaturists). What conformation does it adopt, and why? This question was posed of me by a student at the end of a recent lecture of mine. Surely, an easy answer to give? Read on …

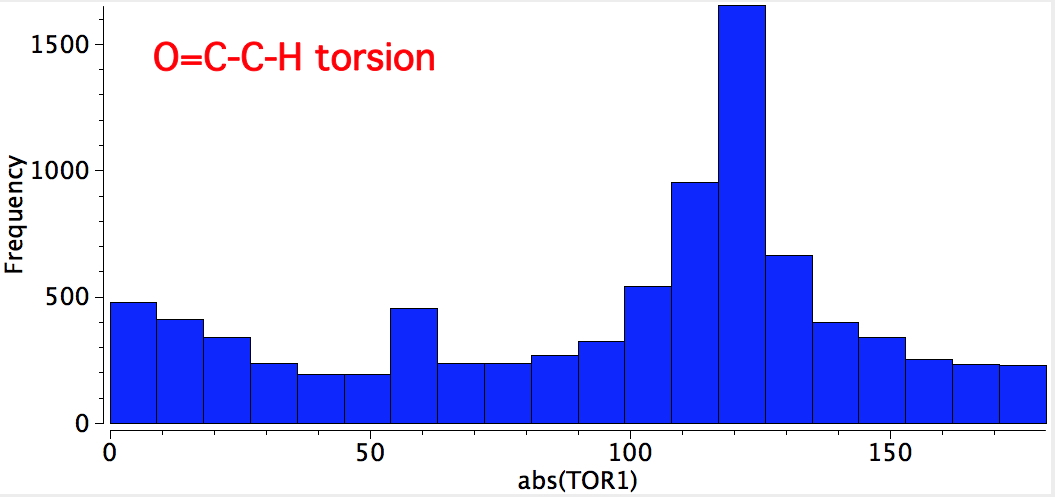
There really are only two possibilities, the syn and anti. Well, I have discovered it is useful to start with a search of the Cambridge data base. With R=H or C, X unspecified, acyclic and T ≤ 175K, two searches were performed. The first identified the torsion around O=C-C-H. This clearly shows a maximum at 120° (with twice the probability), and a smaller one at 0°. This matches syn; the anti conformation above would be expected to have peaks at 60° and 180°; the latter in particular is singularly missing.
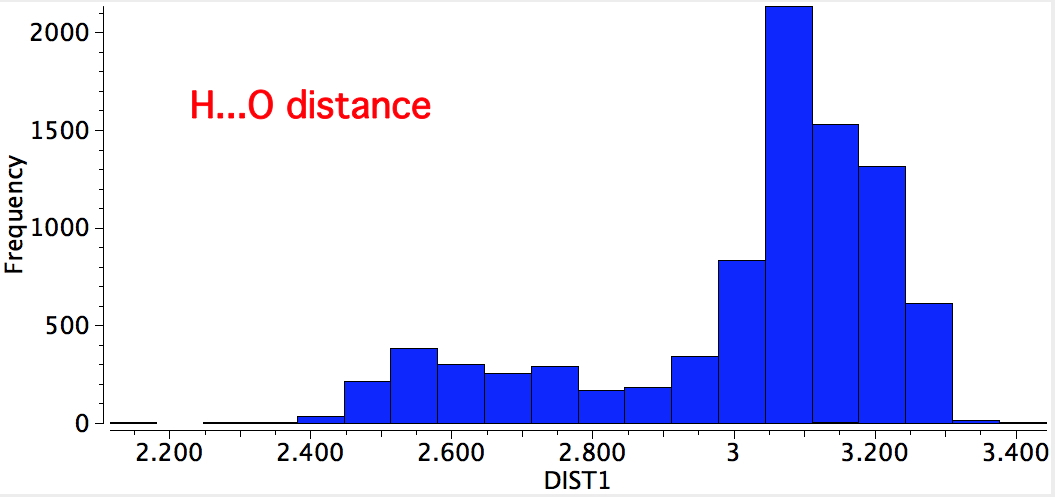
An alternative search is to define the distance between the oxygen and the H. For the syn conformer, distances of ~2.5 and 3.1Å are expected; for the anti conformer, 2.7 and 3.3Å. Again, syn matches better. Remember, searches based on the position of a hydrogen are less reliable than most, so these distributions provide only a statistical indication.
Now for a (ωB97XD/6-311G(d,p) calculation of the rotational barrier. The minima occur at torsions of 0, 120 and 240°, matching syn, although the barrier is very low.

Now to try to find explanations. The standard one finds this in three effects:
- Donation from two C-H bonds (R=H above) into the π*C=O NBO orbital (in the manner that was used to explain the cis-orientation of the two methyl groups in cis-butene).
- Donation from the single co-planar C-H bond into the σ*C=O NBO orbital (blue bonds above)
- Pauli bond-bond repulsions between two filled NBOs.
Effect 1 has an NBO perturbation energy E(2) of 7.0 kcal/mol for the syn conformer and 6.45 for the anti. The explanation is the π*C=O NBO "leans outward", overlapping better with the C-H bonds in the syn than in the anti. the One up to the syn! Effect 2 has values of 1.3 for the syn and 4.1 for the anti. The latter now has the edge. But wait, there are other (smaller) interactions. The syn has an antiperiplanar orientation of the two C-H bonds shown above (X=H,red), E(2) = 3.3 vs 0.6 for the corresponding syn-planar orientation in the anti-conformation. It's now a tie; neck-and-neck.
Effect three suggests that the disjoint NLMO steric exchange energy is 54.34 for the anti and 53.88 (i.e. lower) for the syn. It is vaguely disappointing that no absolutely clear-cut explanation emerges. But then the difference (in total free energy) is only 1.4 kcal/mol. But even this small difference in energy can manifest in fairly clear-cut conformational preferences obtained from crystal structures. Ultimately of course, all effects in chemistry are reducible to the sum of lots of small effects (in other words unpredictable until one does the sum).
I cannot end without mentioning the largest of all the NBO interactions, namely the in-plane lone pair on the oxygen as donor and the aldehyde proton C-H as acceptor (X=H). This has values of 29.3 for syn and 28.8 kcal/mol for anti. This manifest (inter alia) in a greatly reduced C-H vibrational wavenumber (ν 2982 for syn, 2900 cm-1 for anti) compared to the methyl C-H values (~3043-3164).
So this tiny little molecule ended up a little less obvious than might have seemed at the outset. One can find interesting things in even the tiniest of things!
HC…C-H alignment. Click for 3D.
O=C*…C-H alignment. Click for 3D.
Acknowledgments
This post has been cross-posted in PDF format at Authorea.
Author
Tags: Cambridge, conformational analysis, energy, free energy, steric exchange energy, Tutorial material
I agree fully that the “real” explanation is complex, but I have pointed out a simple mnemonic, based on Pople’s early Fourier dissection of barrier contributions, to my students for nearly 40 years: Whatever the reason, rotational barriers (in kcal/mol) of simple systems depend on the number of eclipsed vicinal bonds in the TS: 1 kcal/mol for O-C in alcohols, 2 for H2N-C in amines, and 3 for H3C-C in alkanes. This also works remarkably well for many derivatives. Double bonds can be regarded as being “bent;” T+hus “staggered” conformations with eclipsed single and vicinal (bent) double bonds are best. Aldehyde rotational TS’s thus have one pair of eclipsed single bonds and a ca. 1 kcal/mol barrier, The barrier in propene also should be 1 (instead of 2) kcal/mol on this basis, but the repulsive 1,3-π antibonding interaction in the TS adds another kcal/mol. Not perfectly correct, but easy to remember.