The electronic interaction between a single bond and an adjacent double bond is often called σ-π-conjugation (an older term for this is hyperconjugation), and the effect is often used to e.g. explain why more highly substituted carbocations are more stable than less substituted ones. This conjugation is more subtle in neutral molecules, but following my use of crystal structures to explore the so-called gauche effect (which originates from σ-σ-conjugation), I thought I would have a go here at seeing what the crystallographic evidence actually is for the σ-π-type.

The basic two molecules are shown above; in effect propene 1 and butene 2. The latter was in fact the topic of another post, in which I attempted to show that the close H…H contact in cis-butene (2.1Å) was in effect an unwelcome consequence of the σ-π-conjugation of any of the four "outward leaning" C-H bonds of the methyl groups acting as donors (red-blue below) overlapping with the similarly "outward leaning" π* orbital of the alkene (purple-orange below; blue and purple overlap positively).
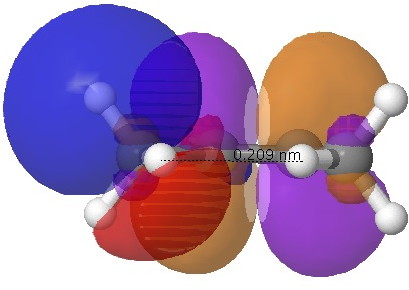
NBO orbitals for C-H/alkene interaction. Click for 3D.
So how general might this be? To find out, I performed the following search on the Cambridge crystal database: 
- The search defines an alkene, bearing two cis-substituents each with at least one C-H bond. The substituents are both sp3 carbon, and the attachment bond to the alkene is defined as acyclic
- The H…H distance uses normalised terminal hydrogen positions (to try to correct for the normally over-short C-H bond lengths found by X-ray).
- Other constraints were R factor < 0.05, no disorder, no errors and (perhaps most importantly) T < 150K to try to reduce thermal libration.
I should qualify all of this by reminding that hydrogen positions in crystal structures are notoriously prone to errors. Nevertheless, with 624 hits using the above search, one might hope for statistical significance of a real effect.
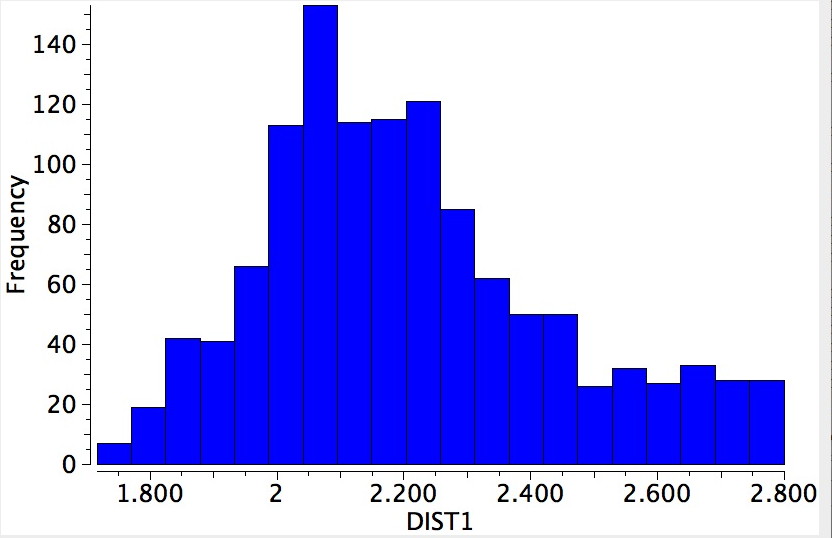
Search result for close H…H contacts in cis-butenes.
For this sample, the most frequent H…H distance emerged as 2.1Å. This can only result from having the C-H bonds lie coplanar with the C=C alkene, as is shown above. The value is also remarkably close to the H…H distance for cis-butene itself (both computationally and as determined using electron diffraction). This does I feel provide a strong indication that σ-π-conjugation is manifesting in these systems.
Re-defining the search for propenes 1 as above gives 1656 hits, with a maximum in the distribution at 2.35Å corresponding to a syn-orientation of the C=C and the C-H bonds. The smaller maximum at about 2.75Å arises from a gauche-orientation between the C=C and C-H (in effect you have to halve this number, since there are twice as many possibilities for this to occur than for the syn). The "inward leaning" gauche C-H bond overlaps less well with the "outward leaning" π* orbital of the alkene.
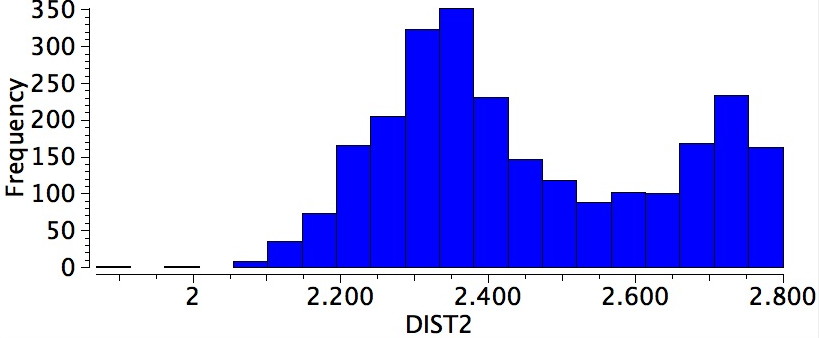
Search result for close H…H contacts in propenes.
These aspects are perhaps better seen in the orbital overlaps shown below.
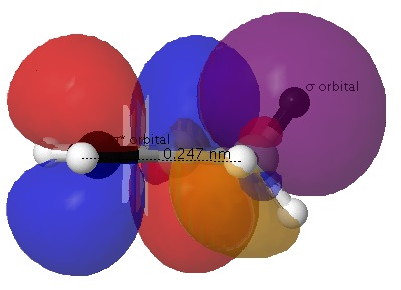
Click for 3D.
I will follow-up this theme with esters and amides next.
Acknowledgments
This post has been cross-posted in PDF format at Authorea.
Author
Tags: above search, Cambridge, conformational analysis, Tutorial material, X-ray
[…] Chemistry with a twist « σ-π-Conjugation: seeking evidence by a survey of crystal structures. […]