A simple correlation between a ring size and the hydrogen bonding as quantified by the O(Lp)/H-O σ* NBO interaction in that ring, indicated a 7- or 8-membered ring was preferred over smaller ones. Here is the same study, but this time using the π-electrons of an alkene as the electron donor.

| n | E(2), kcal/mol | O…H length, Å | Angle,° |
|---|---|---|---|
| 2 | 0.84 | 2.54, 2.75 | 107.4, 126.7 |
| 3 | 1.37 | 2.35, 2.92 | 133.1, 152.9 |
| 4 | 2.04 | 2.45, 2.63 | 139.1, 144.5 |
| 5 | 1.89 | 2.56, 2.63 | 152.2, 138.9 |
The E(2) interaction energies between the NBO filled π-orbital localised to the alkene and the H-O σ* are significantly smaller than for O(Lp) as donors. The energy gap between the donor and acceptor orbitals is smaller for π (ΔE=0.919) than for the O…HO (0.969 au) and if one can compare these values across different localisations, then the finger must point to reduced overlap as the more probable explanation.
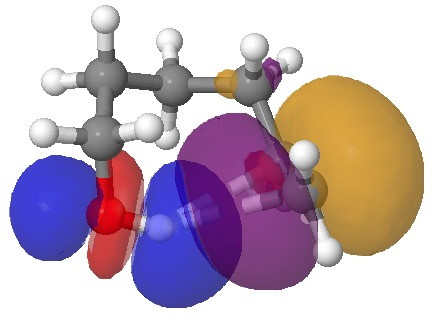
NBO interaction for 7-ring H-bond. Click for 3D.
So does that mean that π bonds do not form? Well take a look at this molecule:[1], which exhibits one example of a π-bond and one of a conventional O…HO bond. E(2) for the π interaction is now 6.6 kcal/mol, and the C…H distance is 2.16Å (calc, 2.1-2.2Å at its shortest, obs).
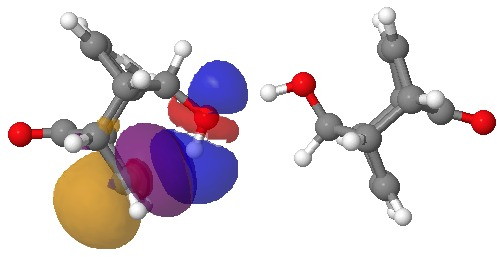
NBO interaction for 7-ring H-bond. Click for 3D.
There are probably several reasons for this.
- The geometry of the scaffold enforces close proximity of the alkene and the OH group. The geometry of the scaffold results in a greater overlap of the C=C π and H-O σ* NBOs. Recollect however the H…H distance in cis-butene, a proximity that was enforced by other effects and which I argued was NOT a chemical bond.
- The energy gap between filled and empty NBOs is slightly reduced from 0.919 to to 0.902 au.
- This in turn may arise because the acceptor strength of the H-O σ* is enhanced by a knock-on effect by an additional conventional hydrogen bond the O (Lp) on this oxygen is forming to a H-O of a second molecular unit.
So we may conclude that whilst strong π H-O hydrogen bonds can be observed, they arise for the molecule shown above from a co-operation of several affects associated with the specific environment, rather than the intrinsic propensity of this type of H-bond to form strong interactions. But one more surprise (perhaps, or possibly just a further insight). The NCI (non-covalent-interactions) surface.
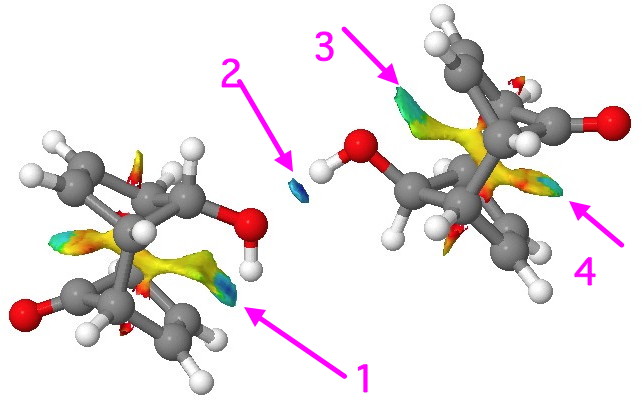
Non-covalent-interactions (NCI) surface. Click for 3D.
This maps the character of the (electron) density gradients, and shows up as blue (attractive) and red (repulsive). Four different regions show up as blue. Arrow 1 is our C=C π…H-O bond. Note how adjacent to the blue region is a green-yellow one, indicating weak repulsions there (this proximity of both attractive and repulsive regions of the density was suggested to account for the confusion surrounding the H…H interaction in cis-butene). Blue on its own however occurs in the region indicated by arrow 2. In this, the conventional O…H-O attraction has no associated repulsion, which may also be an explanation for why such bonds are far more common. But the surprise is arrow 3, which suggests attractive back donation from the O(Lp) to the π* empty orbital (E(2) 1.4 kcal/mol) and arrow 4 indicates an attractive donation from the C=C π to the C=O π* (E(2) = 0.6 kcal/mol). In this particularly rigid system, all these are largely enforced by the atom-scaffold. but it does all show how we often miss noticing the weaker interactions in molecules.
Author
References
- M. Etzkorn, S.D. Smeltz-Zapata, T.B. Meyers, X. Yu, and M. Gerken, "From the anti-tricyclo[4.2.1.12,5]deca-3,7-diene framework to 4,5,6,7-tetrachloro-isoindenone derivatives", Tetrahedron Letters, vol. 51, pp. 6075-6077, 2010. https://doi.org/10.1016/j.tetlet.2010.09.045
Tags: energy gap
Such intramolecular OH–π hydrogen bonding was one of my first research projects as a young instructor at Princeton: (Intramolecular Hydrogen Bonding Involving Double Bonds, Triple Bonds, and Cyclopropane Rings as Proton Acceptors. Paul von Rague Schleyer, Daniel S. Trifan, and Robert Bacskai. J. Am. Chem. Soc. 1958, 80, 6691- 6692. Also see the analogous phenyl cases: Hydrogen Bonding in ß-Aryl Ethanols. Paul von Rague Schleyer, C. Winter, D.S. Trifan, and R. Bacskai. Tetrahedron Lett. 1959, 1-7. The IR detection used left no doubt about the H-bonding, due to the characteristic separate frequency-shifted OH…π peaks. Many such OH…π examples were reported subsequently.
Thanks Paul. Yes, together with C-H…O interactions, such hydrogen bonds are often under-rated. A conventional O…H hydrogen bond originates from a two-electron donor. What makes the π-electron variety so interesting is that examples are known with 2, 4 and 6 electrons as donors.
I do wonder whether any examples of > 6 might exist?
[…] In this post, I looked at some hydrogen bonds formed by interaction of a π-system with an acidic hydrogen. Unlike normal lone pair donors, π-systems can involve more than two electrons, most commonly four or six. Here I look at examples of both these higher-order donors. […]
[…] in at around 3 kcal/mol. So this one is very weak. This is due to poor overlap resulting from the small ring size […]