tpap[1], as it is affectionately known, is a ruthenium-based oxidant of primary alcohols to aldehydes discovered by Griffith and Ley. Whereas ruthenium tetroxide (RuO4) is a voracious oxidant[2], its radical anion countered by a tetra-propylammonium cation is considered a more moderate animal[3]. In this post, I want to try to use quantum mechanically derived energies as a pathfinder for exploring what might be going on (or a reality-check if you like).

A basic (i.e. simple) mechanism for oxidation of an alcohol by RuO4 is shown above. Here I reality-check this mechanistic pathway with the help of ωB97XD/Def2-SVPP/SCRF=dichloromethane calculations. I should point out that since the mechanism is going to involve ion-pairs, it is particularly important to adopt a solvent=corrected model from the outset[4]. TS1 is the transition state for addition of the alcohol to the metal, a process which involves a synchronous proton transfer for the singlet electronic state.
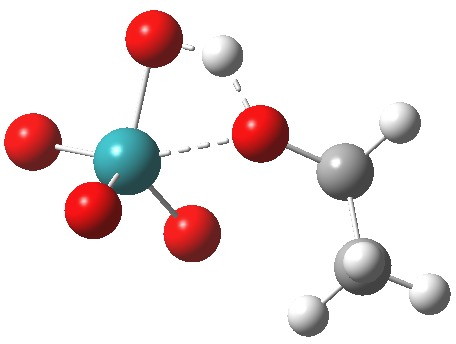 TS1 as a singlet. Click for 3D |
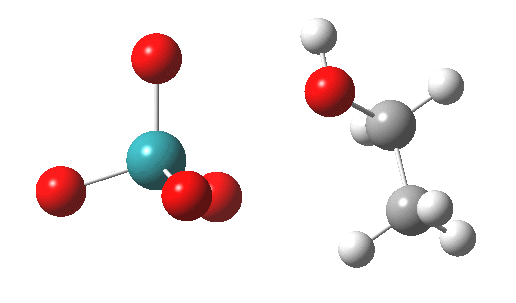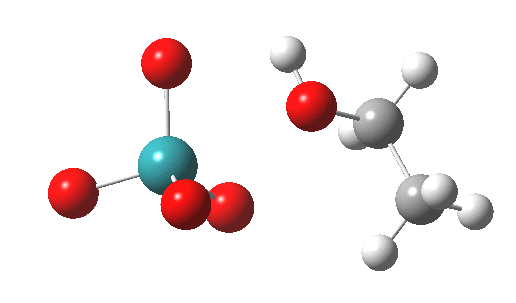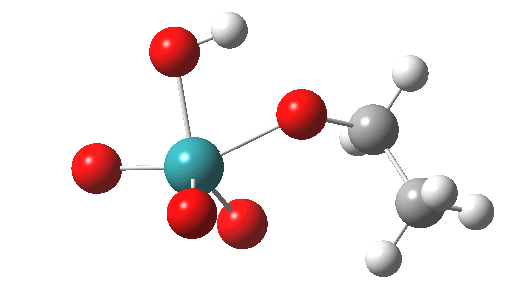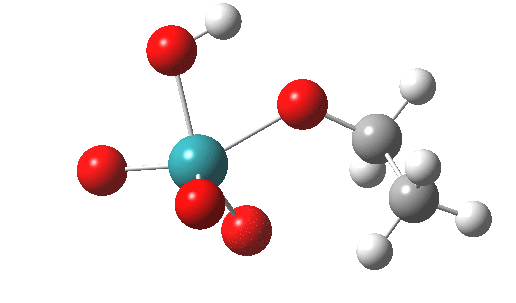 |
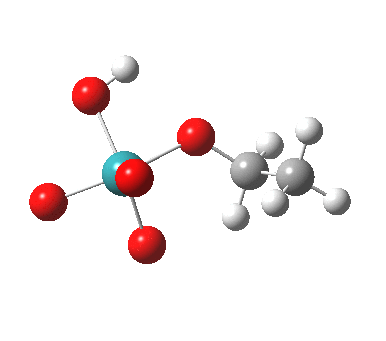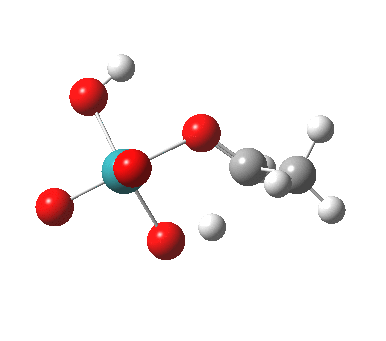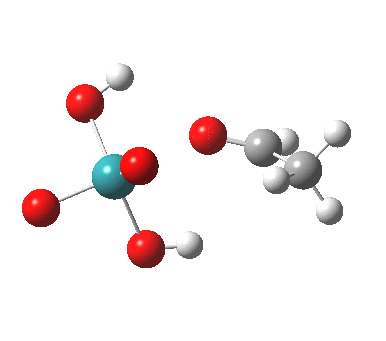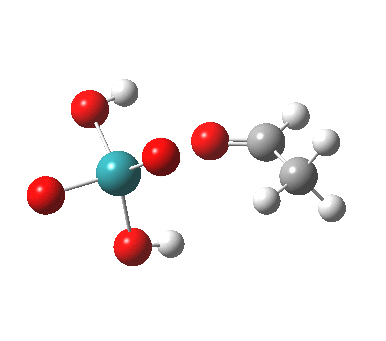
Next comes TS2, which involves a hydride abstraction with concomitant reduction of the oxidation number of Ru(VIII) to Ru(VI). It is higher in free energy than TS1 by 1.1 kcal/mol. The barrier corresponds to ΔG298‡ 37.1 kcal/mol. The process completes by low energy elimination of water (TS3) from the Ru(VI) species to give RuO3, which either undertakes further oxidisation to give RuO2, or might instead be re-oxidized back to RuO4 by oxygen (or an amine N-oxide) to complete a catalytic cycle.
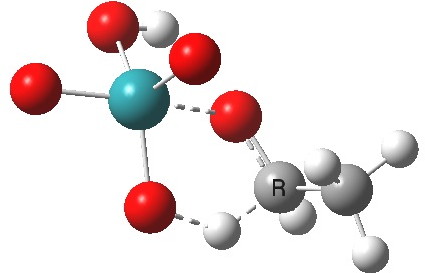 TS2 as a singlet. Click for 3D |
–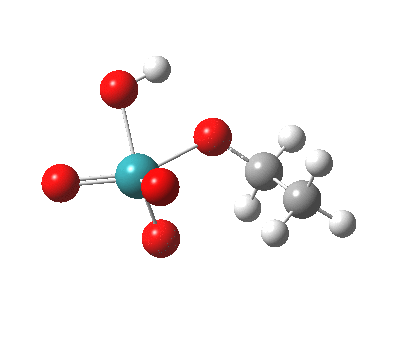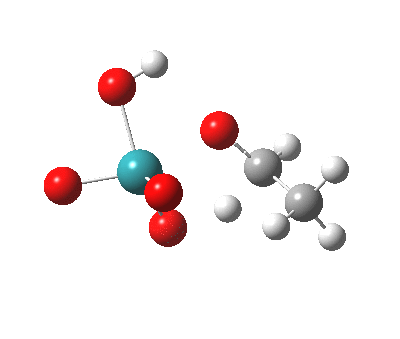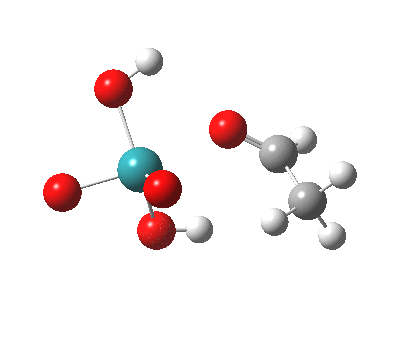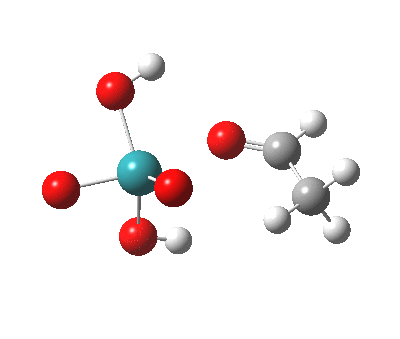 |
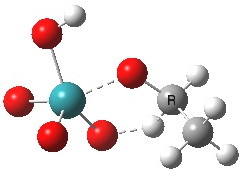 TS2 as a triplet. Click for 3D |
 |
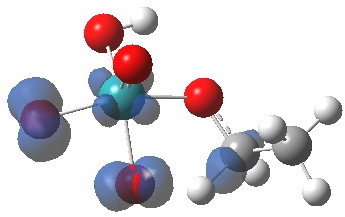
Right away, we have a problem; ΔG298‡ 37.1 kcal/mol is too high to be a realistic pathway, and yet RuO4 is a known oxidant[2]. One way out is to see if the triplet state energy of this system might be lower. Whilst the triplet-state reactant is higher in energy (by 27.5 kcal/mol) , TS2 is lower and corresponds to a reduced barrier of ΔG298‡ 28.2 kcal/mol. Better, but a (small?) question mark still remains, since one would really expect the barrier to be ~20 kcal/mol or less for a “voracious oxidant”. Perhaps the incursion of triplets makes it indiscriminate? The spin density at the transition state is shown below, it extends across both oxygen, carbon and Ru.
The tpap modification to this process is to use Ru(VII) in the form of a radical anion partnered with a quaternary ammonium cation. The basic reagent is therefore an ion-pair, hence the solvation approach mentioned earlier is needed to describe the energetics of such a species. The R alkyl groups here are modelled as methyl rather than propyl.

TS2 for this radical-ion-pair is shown below, and it has ΔG298‡ 30.8 kcal/mol, 2.6 kcal/mol higher than for the un-moderated reagent. In this case, the higher-spin quartet states are higher in energy (by at least ~7.9 kcal/mol) and so do not participate.
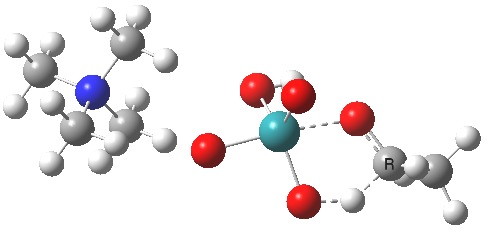 |
The spin density for tpap-TS2 also reveals it to be concentrated on Ru and one oxygen. Little is transferred to the ethanol, and we might infer then that this TS corresponds to transfer of two-electrons from the ethanol to the Ru-oxidant. This corresponds to Ru(VII) being reduced to Ru(V), i.e. a 2-electron oxidation/reduction. Unfortunately, an attempt to chart the reaction across a whole reaction coordinate (IRC) failed with SCF-convergence problems, which might suggest a change in the spin-configuration during this process. A more sophisticated multi-configurational approach might be needed to properly establish the electron dynamics of what is turning out to be a more complex reaction than first seemed.
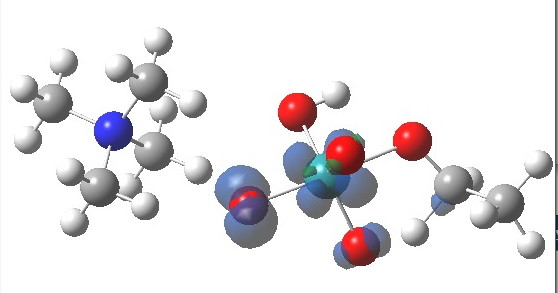
It is time to sum up what might have been learnt.
- A reality check on the energetics of a viable-looking mechanistic route can establish whether such a mechanism does have a low enough free-energy to be viable at (in this case) room temperatures.
- In fact, observing that our initial mechanism had too high an energy led us to discover a triplet-state path that was significantly lower in energy. However, even this is still a bit too high.
- The tpap-variation of the oxidant, which enforces a doublet-state upon the mechanism,has a barrier which appears to be similar to the triplet-state RuO4 mechanism. This too may be too high in energy. At least we can probably rule out a quartet-state mechanism.
- And so it seems appropriate to end here by noting that experimentally[3] the kinetics of tpap oxidations appear to be autocatalytic. The rate speeds up once some RuO3 (or RuO2) has been formed, and this suggests that perhaps a binuclear system containing two Ru atoms is a faster oxidant than the mononuclear variety. This reminds of the mechanism for Sharpless perepoxidation, where two metal centres were needed to control the stereochemistry.
So after all of this, we have not really found an explanation of why tpap is a more selective and moderate oxidant than the rapacious RuO4. But perhaps this is because more complex models with more than one Ru-atom need to be constructed. This would in turn allow the oxidative hydride abstraction from the alcohol to occur in a larger (7) ring transition state, which is always the preferred geometry for such transfers. If I find such, I will report back here.
Author
References
- S.V. Ley, J. Norman, W.P. Griffith, and S.P. Marsden, "Tetrapropylammonium Perruthenate, Pr<sub>4</sub>N<sup>+</sup>RuO<sub>4</sub> <sup>-</sup>, TPAP: A Catalytic Oxidant for Organic Synthesis", Synthesis, vol. 1994, pp. 639-666, 1994. https://doi.org/10.1055/s-1994-25538
- D.G. Lee, U.A. Spitzer, J. Cleland, and M.E. Olson, "The oxidation of cyclobutanol by ruthenium tetroxide and sodium ruthenate", Canadian Journal of Chemistry, vol. 54, pp. 2124-2126, 1976. https://doi.org/10.1139/v76-304
- D.G. Lee, Z. Wang, and W.D. Chandler, "Autocatalysis during the reduction of tetra-n-propylammonium perruthenate by 2-propanol", The Journal of Organic Chemistry, vol. 57, pp. 3276-3277, 1992. https://doi.org/10.1021/jo00038a009
- J. Kong, P.V.R. Schleyer, and H.S. Rzepa, "Successful Computational Modeling of Isobornyl Chloride Ion-Pair Mechanisms", The Journal of Organic Chemistry, vol. 75, pp. 5164-5169, 2010. https://doi.org/10.1021/jo100920e
Tags: catalysis, energy, free energy, low energy elimination, metal, react freq, Reaction Mechanism, RuO4+ ethanol, triplet state energy, Tutorial material
See, DOI: 10.1002/ejoc.201800860, where this blog post is referenced. A definite explanation is not (yet) presented either, it seems. They consider an outer-sphere mechanism via H-bonds only.
Thanks for tracking this. It is always nice to see that other forms of “publication” can achieve citations!