Here is an inside peek at another one of Derek Lowe’s 250 milestones in chemistry, the polymorphism of Ritonavir.[1] The story in a nutshell concerns one of a pharma company’s worst nightmares; a drug which has been successfully brought to market unexpectedly “changes” after a few years on market to a less effective form (or to use the drug term, formulation). This can happen via a phenomenon known as polymorphism, where the crystalline structure of a molecule can have more than one form.[2],[3],[4] In this case, form I was formulated into soluble tablets for oral intake. During later manufacturing, a new less-soluble form appeared and “within weeks this new polymorph began to appear throughout both the bulk drug and formulation areas“[1]
The structure of the original form I is shown below (3D DOI: 10.5517/CCRVC75). The compound has three HN-CO peptide linkages, all of which are in the stereoelectronically favoured s-cis form, with a dihedral angle of 180° across the H-N and C=O vectors.
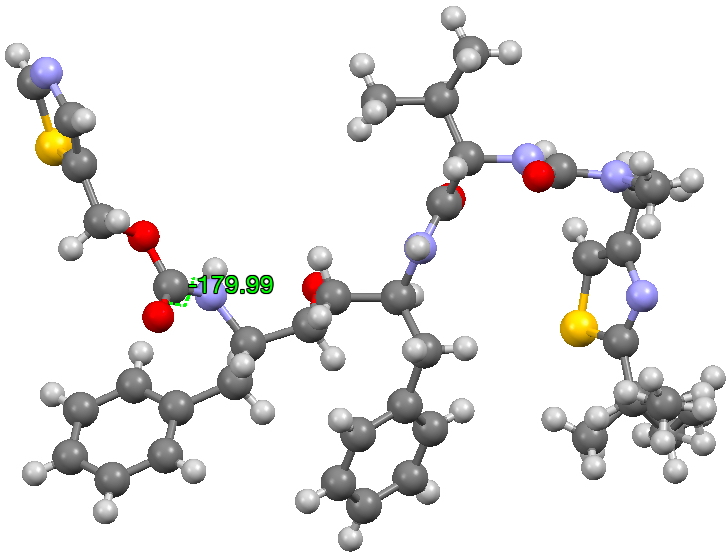
Click for 3D
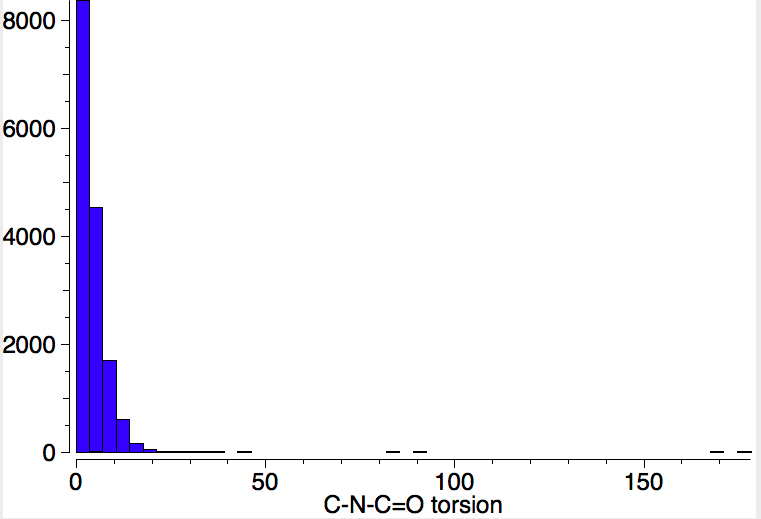
To show how favourable this s-cis form is, here is a search of the Cambridge structural database for acyclic HN-C=O bonds; of the ~8200 examples, only 5 have an s-trans torsion of ~180°. It is I feel statistically not entirely correct to convert this ratio of K=1640 to a free energy, but if one does, then at 298K, RTlnK works out to 4.4 kcal/mol. Note also that two compounds show an angle of ~90° (artefacts?).
The new type-II form that emerged has only two s-cis peptide linkages, and the third has isomerised to this higher energy s-trans form (3D DOI: 10.5517/CCRVC97)
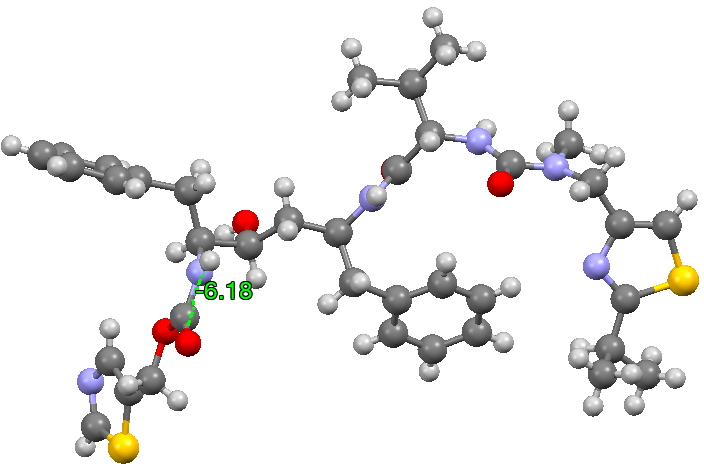
Click for 3D
This has various knock-on effects on the conformation of the actual molecule itself.
- The cis-trans isomerisation of a peptide or amide bond is a relatively high energy process, since the C=N bond order is higher than 1. For example, in the 1H NMR spectrum of N,N-dimethyl formamide at room temperature, one can famously observe two methyl resonances and it is only at higher temperatures that the two signals coalesce due to more rapid rotation about the C=N bond.
- A pedant might query whether this isomerism is correctly termed a conformational or a configurational change? High-energy rotations that result in cis/trans isomerisms are normally referred to as a configurational changes, whereas low energy rotations about e.g. single bonds are known as conformational changes (thus the conformational changes in cyclohexane). There is a grey region such as this one, where the boundary between the two terms is encountered.
- This isomerism has the knock-on effect of inducing a much lower energy rotation of a C-C single bond (on the left hand side of the representations above), rotating from a dihedral angle of +193 in form I to +51 in form II.
- More minor affects are seen in the conformation of the central benzyl group and the S/N heterocyclic ring on the right hand side.
- All these low energy conformational effects occur because a better hydrogen bonding network can then be set up in the crystal lattice, something not easily predictable from the diagrams of the single molecules shown above.
- Overall, the free energy of the lattice is lower, despite the higher energy of the s-trans peptide bond.
- Clearly, the dynamics of crystallisation initially favoured form I (despite the higher energy of the crystallised outcome), but if a tiny seed of form II is present (or perhaps other impurities) this can dramatically (but unpredictably) change these crystallisation dynamics.
I suspect that since 1998 when this story unfolded, all new drugs in which one or more s-cis peptide bonds are present have caused anxiety. In the system above for example, one might ask whether cis/trans isomerisation of instead either of the other two peptide bonds present might have similar results? Or hypothesize whether inhibiting the associated rotation of the C-C single bond noted above by appropriate “tethering” might prevent form I from converting to form II. Since 1998, I am sure trying to predict the solid form of an organic molecule from its isolated structure using computational methods has dramatically increased, although I have not found in SciFinder any reported instances of such modelling for Ritonavir itself.[5] Perhaps, if such a method were found, it might be too commercially valuable to share?
Author
References
- J. Bauer, S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter, and J. Morris, "Ritonavir: An Extraordinary Example of Conformational Polymorphism", Pharmaceutical Research, vol. 18, pp. 859-866, 2001. https://doi.org/10.1023/a:1011052932607
- J.D. Dunitz, and J. Bernstein, "Disappearing Polymorphs", Accounts of Chemical Research, vol. 28, pp. 193-200, 1995. https://doi.org/10.1021/ar00052a005
- D. Bučar, R.W. Lancaster, and J. Bernstein, "Disappearing Polymorphs Revisited", Angewandte Chemie International Edition, vol. 54, pp. 6972-6993, 2015. https://doi.org/10.1002/anie.201410356
- G.J.O. Beran, I.J. Sugden, C. Greenwell, D.H. Bowskill, C.C. Pantelides, and C.S. Adjiman, "How many more polymorphs of ROY remain undiscovered", Chemical Science, vol. 13, pp. 1288-1297, 2022. https://doi.org/10.1039/d1sc06074k
- S.R. Chemburkar, J. Bauer, K. Deming, H. Spiwek, K. Patel, J. Morris, R. Henry, S. Spanton, W. Dziki, W. Porter, J. Quick, P. Bauer, J. Donaubauer, B.A. Narayanan, M. Soldani, D. Riley, and K. McFarland, "Dealing with the Impact of Ritonavir Polymorphs on the Late Stages of Bulk Drug Process Development", Organic Process Research & Development, vol. 4, pp. 413-417, 2000. https://doi.org/10.1021/op000023y
Tags: Carbamates, Chemistry, Derek Lowe, free energy, high energy process, High-energy rotations, higher energy, higher energy s-trans form, hydrogen bonding network, later manufacturing, Lipid polymorphism, low energy conformational effects, low energy rotations, lower energy rotation, Peek, Polymorphism, Protease inhibitors, Ritonavir, RTT, SN, Software engineering, Thiazoles, Ureas