A project fork is defined (in computing) as creating a distinct and separate strand from an existing (coding) project. Here I apply the principle to the polar azulene 4 explored in an earlier post, taking m-benzyne as a lower homologue of azulene as my starting point.

m-Benzyne is a less stable 1,3 isomer of o-benzyne (1,2-dehydrobenzene), and is often represented as a 1,3-biradical of 1,3-dehydrobenzene. But, could it be stabilized with cyano and amino groups as shown in 5 above? Here the idea is that charge transfer from the 3-ring to the 5-ring will create a lower homologue of azulene (a well known molecule), with the 3-ring a 4n+2 π-electron aromatic (n=0) and the five ring similarly so (n=1).
I start with the computed (wB97XD/Def2-TZVPP/SCRF=thf) structure of m-benzyne itself, as a closed shell molecule (DOI: 10.14469/hpc/1995). The C-C bond connecting the two rings is long (with a biradical tendency) and hence the conjugation is restricted to the outer periphery. The dipole moment is 0.51D (the dipole vector as shown in blue has the expected direction of polarity).
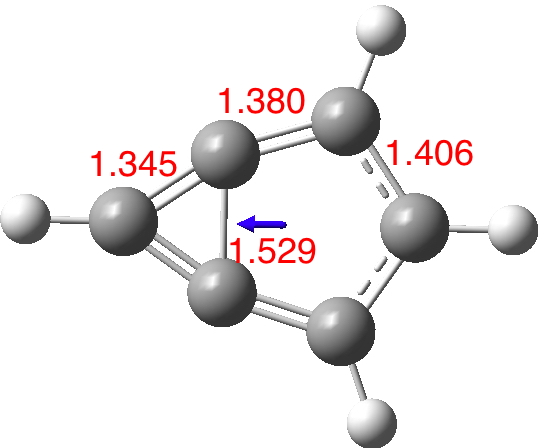
Now compare this to the substituted version 5; the bond lengths are all more characteristic of aromatic values and most significantly the central bond is as well (DOI: 10.14469/hpc/1996). The dipole moment is augmented thirty fold to 14.6D, which would rank alongside that reported for the most polar neutral molecule.
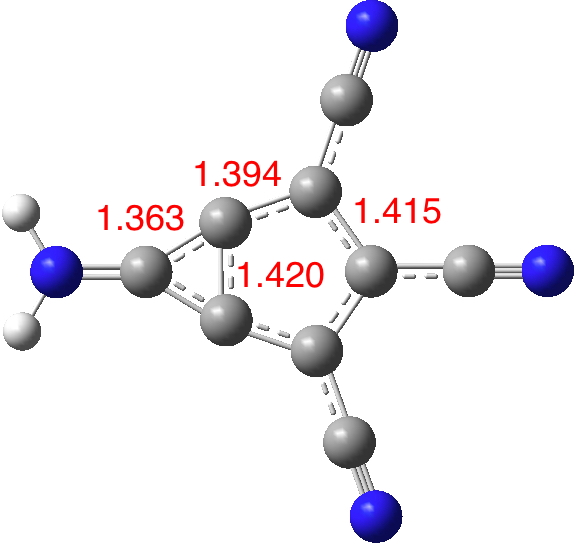
So I suggest this is substituted “m-benzyne” well worth trying to make and one very much unlikely to have any dispute about the nature of its wavefunction, i.e. biradical or closed shell.
Author
Tags: Aryne, Azulene, Chemical polarity, Chemistry, Dipole, Extreme umpolung
The system described above has 6 π-electrons, partitioned to sit on the five membered ring allowing it to become aromatic. What would happen to an 8π analogue, as below (1,3-dehydrocyclo-octatetraene).
Inducing all 8 π-electrons to sit on the seven membered part could achieve two results.
1. The 7-ring becomes antiaromatic, hardly a driving force for charge transfer one might think!
2. The 7-ring could twist to become Möbius aromatic, perhaps a better driving force, but offset by the strain induced by twisting.
The calculation (DOI: 10.14469/hpc/1997) reveals a planar molecule with no negative force constants, but a dipole moment of 16.6D (the vector in blue is in the usual direction of -ve to +ve).
The NICS(1) value at the 1Å above the centre of the 7-ring is +37 ppm, which is highly antiaromatic. So, amazingly, this molecule prefers to become antiaromatic in its quest to become polar. I am sure this cannot be the whole story however (?).
Notice how transposing the substituents on the above system reverses the dipole moment to 14.7D in the opposite direction, that is a change of 31.3D induced by the substituents (DOI: 10.14469/hpc/1999). So there is another record to try to beat; that of extreme umpolung.
The 3-ring is now forced to be antiaromatic, although it avoids this by lengthening the C-C bonds and making the anionic carbon non-planar, in the manner described here. The 7-ring is however aromatic.
Really interesting topic Henry! The m-benzyne analogue would be a fantastic test case for creating this strained bicyclic. Given its high symmetry it should be amenable to CASSCF, CI and CC computations to test the nature of the wavefunction. It would be interesting to see a PES defined by the distance between the two (formal) radical carbon atoms.
Other substituents might be interesting too – how about nitro groups instead of the nitriles?
Steve,
Yes, a multi-reference study would be well worthwhile. Nitro groups introduce a little more complexity; can they all be co-planar? (this issue does not arise with cyano of course). And perhaps CF3 groups as well?