The concept of kinetic vs thermodynamic control of a reaction is often taught in the context of the enolisation of e.g. 1-methylcyclohexanone as induced by a base. The story goes that at low temperatures (-78°C), the rate of the sterically more hindered thermodynamic enolisation does not compete with the faster kinetic product but that at higher temperatures when an equilibrium is possible, the thermodynamically more stable tetrasubstituted enol is formed. I set out to see if this result can be modelled.

I started with the scheme shown above, R=X=H (i.e. two water molecules inducing the enolisation).
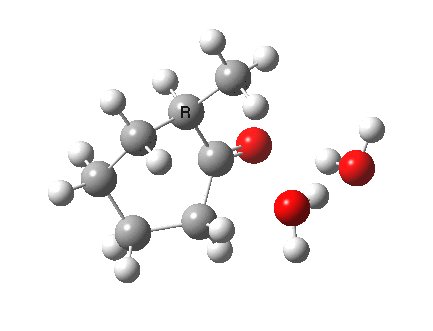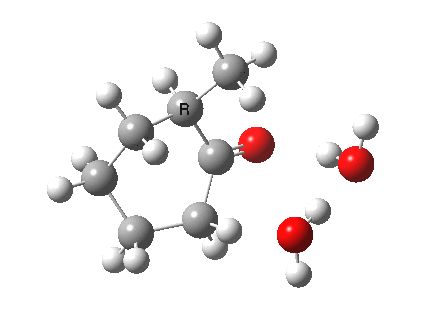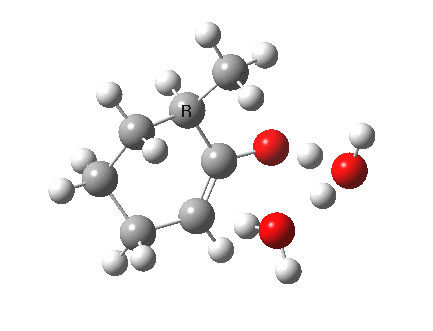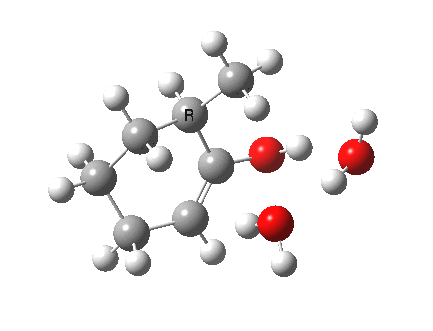 |
|
 |
 |
Points to note:
- The transition state is very late (i.e. product, or enol-like). The cleaving C-H and the forming H-O (base) bond lengths are respectively 1.724 and 1.042Å (ωB97XD/6-311G(d,p)/SCRF=acetone).
- The activation barrier is high (~33 kcal/mol). This is because water is a very weak base.
- The difference in ΔG298‡ shows the thermodynamic[1] transition state is 0.6 kcal/mol lower than the kinetic[2]
- OK, what about ΔG220‡ then? The difference is still 0.5 kcal/mol.
- The reaction is endothermic by about 7 kcal/mol. It is known that enols are less stable than ketones.
- The tetrasubstituted “thermodynamic” enol product is 1.7 kcal/mol more stable than the trisubstituted “kinetic” form.
So, to avoid the transition state inheriting too much of the product-stability, it has to be an early one rather than a late one! How about guanidine as a stronger base[3]? The C-H and H-N lengths are now 1.414 and 1.291Å, and the barrier goes down to ~16 kcal/mol. The transition state is certainly more reactant like, and indeed the ΔG298‡ difference is now 0.0 kcal/mol! Notice also that with the stronger base, the initial product is in fact an ion-pair, comprising the guanidinium cation and the enolate anion.

Something more is clearly needed in the model to convert the kinetic enol to a low-temperature winner. A stronger base probably, but perhaps also a hindered base? If I find such a model, I will report back here.
Postscript: My closing remark above was that I was going to try to locate transition states using a stronger, perhaps more hindered base. The obvious one to try is LDA (lithium di-isopropylamide, or LiNiPr2). This indeed results in an earlier and (possibly) more hindered transition state[4] (C-H 1.227Å). ΔΔG298‡ now is 2.4 kcal/mol in favour of the kinetic over the thermodynamic[5] isomer, which is enough to ensure a ratio of ~60:1 in favour of that outcome at 298K, or ~250:1 at -78°C. The activation barrier is now really very small (~4 kcal/mol) which matches the supposition that it is a very early transition state indeed.
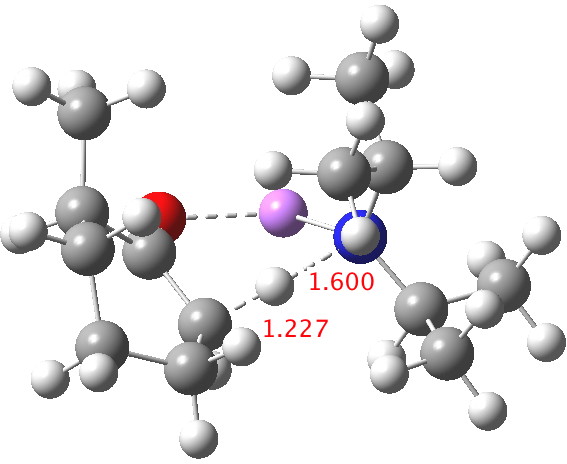
Click for 3D
An NCI (non-covalent-interactions) analysis of the kinetic transition state is shown below. It shows distinct (green = weakly stabilising) zones between the isopropyl groups of the base and the cyclohexanone. So the effect might not be actually “steric-hindrance”, but in fact originate from optimal (dispersion) attractions (shown in blue to green, whilst destabilising steric hindrance would be shown in yellow to red) directing the formation of the kinetic isomer[6].
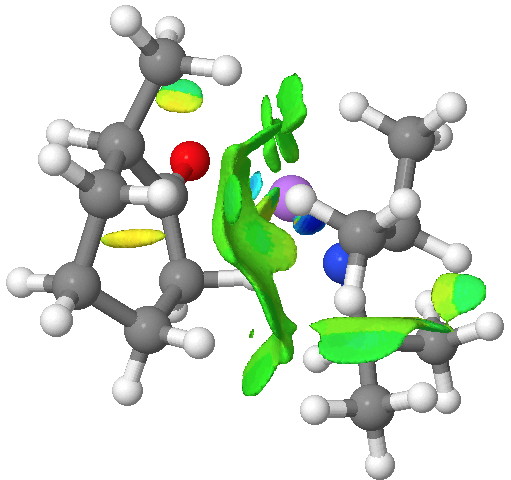
Click for 3D
Li is a complex element, and of course oligomeric structures abound. I am currently exploring these, and if I find anything interesting, there will no doubt be a postscript to the postscript.
Postscript to the postscript. The next model up includes adding one thf as coordinating solvent to the Li. ΔΔG220‡ is now attenuated to 1.1 kcal/mol in favour of the kinetic[7] over the thermodynamic,[8] leading to a 12:1 ratio at 220K.
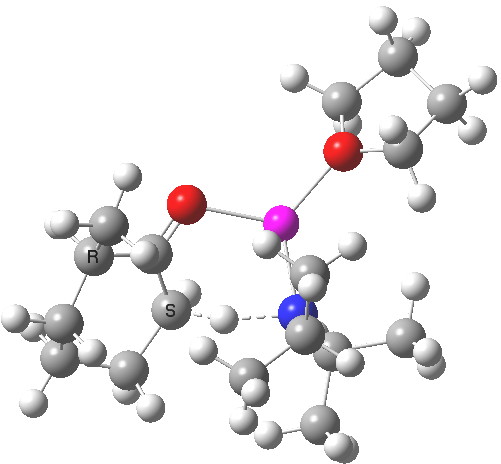
Click for 3D
Author
References
- H.S. Rzepa, "Gaussian Job Archive for C7H16O3", 2013. https://doi.org/10.6084/m9.figshare.840464
- H.S. Rzepa, "Gaussian Job Archive for C7H16O3", 2013. https://doi.org/10.6084/m9.figshare.840465
- H.S. Rzepa, "Gaussian Job Archive for C8H17N3O", 2013. https://doi.org/10.6084/m9.figshare.840490
- H.S. Rzepa, "Gaussian Job Archive for C13H26LiNO", 2013. https://doi.org/10.6084/m9.figshare.841757
- H.S. Rzepa, "Gaussian Job Archive for C13H26LiNO", 2013. https://doi.org/10.6084/m9.figshare.842572
- S. Ehrlich, H.F. Bettinger, and S. Grimme, "Dispersion‐Driven Conformational Isomerism in σ‐Bonded Dimers of Larger Acenes", Angewandte Chemie International Edition, vol. 52, pp. 10892-10895, 2013. https://doi.org/10.1002/anie.201304674
- H.S. Rzepa, "Gaussian Job Archive for C17H34LiNO2", 2013. https://doi.org/10.6084/m9.figshare.845578
- H.S. Rzepa, "Gaussian Job Archive for C17H34LiNO2", 2013. https://doi.org/10.6084/m9.figshare.845579
Tags: faster kinetic product
I have added a postscript above, showing LDA as base in the reaction.
Below is the IRC for this reaction. Particularly noteworthy is the observation that whereas the conversion of ketone to enol by water was endothermic, converting the ketone to the lithium enolate is strongly exothermic! Because of the large reverse barrier, the reaction is not in equilibrium at low temperatures. Note also the small forward barrier, as noted in the post, which is how the reaction at -78°C is rendered regiospecific.
See postscript to postscript, where results for a model which includes one one thf solvent molecule coordinated to the Li. The kinetic transition state is still lower, by 1.1 kcal/mol in free energy, which leads to 12:1 in favour of the kinetic outcome at 220K.