A reader asked me about the mechanism of the reaction of 2-picoline N-oxide with acetic anhydride to give 2-acetoxymethylpyridine (the Boekelheide Rearrangement[1]). He wrote ” I don’t understand why the system should prefer to go via fragmentation-recombination (… the evidence being that oxygen labelling shows scrambling) when there is an easy concerted pathway available (… a [3,3]sigmatropic shift). Furthermore, is it possible for two pathways to co-exist?” Here is how computation might enlighten us.

The first model I built discards the apparently extraneous product in the first reaction, ethanoic acid. A transition state is located (ωB97XD/6-311G(d,p)/SCRF=dichloromethane) and its intrinsic reaction coordinate is shown below.[2]
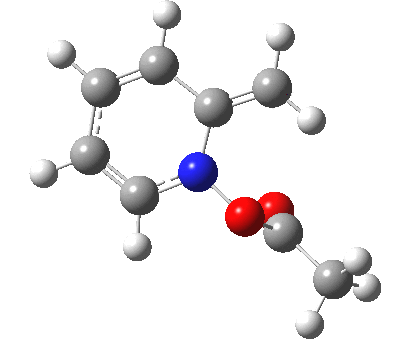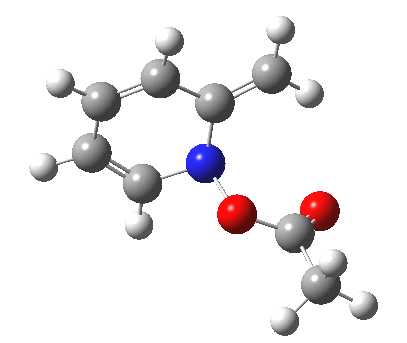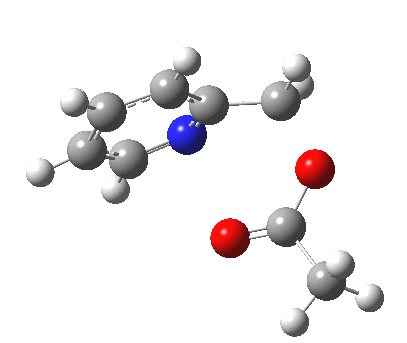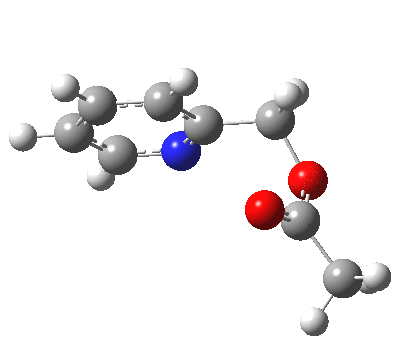
 |
 |
- One first notes that the reaction is concerted, with no intermediates along the route.
- The reaction barrier (~21 kcal/mol) is quite reasonable for a [3,3] sigmatropic reaction.
- There is an almost undiscernible blip (inflexion) in the gradient norm at about +1 and a more obvious one at IRC +8. The latter is a hidden intermediate corresponding to a conformational rotation about the newly formed C-O bond. The former is more significant, since it is providing the faintest of hints that a hidden intermediate[3] corresponding to an ion-pair (in red in the scheme above) might be attempting to form. But it is only a hint, no more.
So an easy concerted pathway is indeed available. But the solvent model (dichloromethane) is not really very polar. How about water, which should better stabilise any ion-pair intermediate? That tiny blip in the gradient norm of the IRC (@~1) becomes a bit more prominent, but the reaction is computed as resolutely concerted.

So to explain why oxygen label scrambling is possible, we have to adopt a better model. That ethanoic acid discarded from our first attempt is re-instated. It serves the purpose of potentially stabilising any ion-pair which might form via explicit hydrogen bonds.[4]
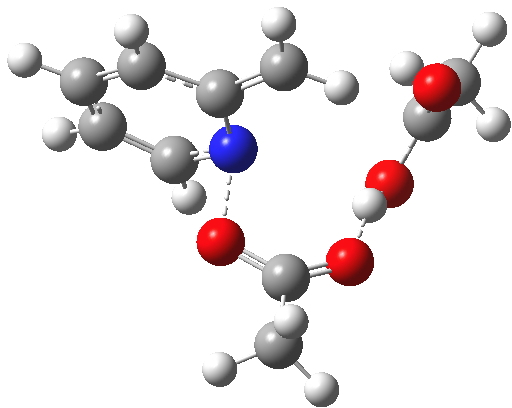
Click for 3D.
The IRC[5] for this variation does indeed show a change; at IRC +3, there is now a very prominent hidden intermediate feature, showing that the additional molecule of ethanoic acid formed in the first step is stabilizing the ion-pair. It also serves to reduce the barrier to the reaction (by ~4 kcal/mol).‡
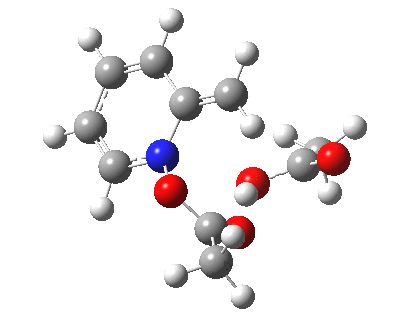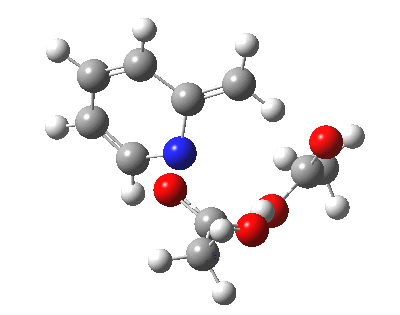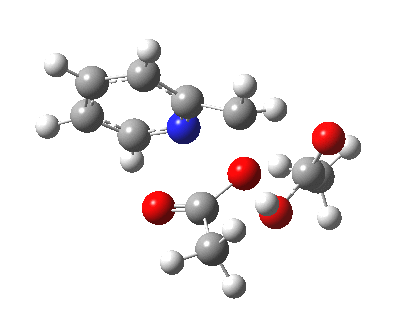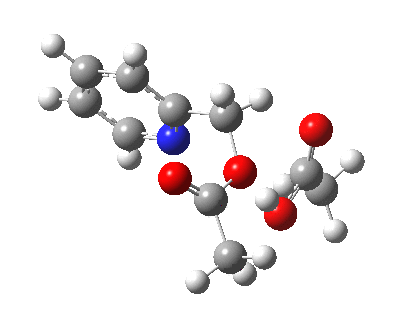 |
|
 |
 |
Although the Boekelheide rearrangement sounds like a rather obscure reaction that few have heard of, discussing it actually introduces an important concept common to many reactions. That is that they can proceed via either relatively neutral or highly ionic pathways, and that the balance between these two may be both subtle and influenced by external factors. In this case, the formation of a hydrogen bond stabilising the transition state for the reaction. This of course is also how many an enzyme achieves its action! For the Boekelheide rearrangement, a single hydrogen bonded ethanoic acid promotes, but does not fully establish the ion-pair mechanism over the neutral [3,3] pericyclic rearrangement. However, one might imagine that adding perhaps a second explicit stabilising H-bond might swing the balance over from merely a hidden intermediate to a real (ion-pair) intermediate. It is also possible that changing the acidity of this component (by replacing e.g. CH3CO2H by e.g. CF3CO2H) might achieve the same result.
As to whether “it is possible for two pathways to co-exist”, a nice example of this in my experience comes from the enantiomerisation of isobornyl chloride in cresol,[6] which has been shown by extensive isotope labelling to proceed by two concurrent but very different pathways. It is probably more common than we realise.
‡ It is worth noting that the [3,3] sigmatropic reaction is unimolecular, whereas the ethanoic-assisted variation is bimolecular. Apart from taking into account the entropic requirements of the latter, it is also necessary to redefine the standard state for the free energy from 1 atm to a more reasonable 1M, which reduces the free energy barrier by about 1.9 kcal/mol, and a correction which reduces the free energy of a bimolecular reaction a further 2.6 kcal/mol can be applied as a solvent correction.[7]. These two corrections mean that bimolecular solution reactions are often not so unfavourable compared to unimolecular equivalents as is often made out.
Author
References
- A. Massaro, A. Mordini, A. Mingardi, J. Klein, and D. Andreotti, "A New Sequential Intramolecular Cyclization Based on the Boekelheide Rearrangement", European Journal of Organic Chemistry, vol. 2011, pp. 271-279, 2010. https://doi.org/10.1002/ejoc.201000936
- H.S. Rzepa, "Gaussian Job Archive for C8H9NO2", 2013. https://doi.org/10.6084/m9.figshare.730627
- E. Kraka, and D. Cremer, "Computational Analysis of the Mechanism of Chemical Reactions in Terms of Reaction Phases: Hidden Intermediates and Hidden Transition States", Accounts of Chemical Research, vol. 43, pp. 591-601, 2010. https://doi.org/10.1021/ar900013p
- H.S. Rzepa, "Gaussian Job Archive for C10H13NO4", 2013. https://doi.org/10.6084/m9.figshare.730621
- H.S. Rzepa, "Gaussian Job Archive for C10H13NO4", 2013. https://doi.org/10.6084/m9.figshare.731688
- J. Kong, P.V.R. Schleyer, and H.S. Rzepa, "Successful Computational Modeling of Isobornyl Chloride Ion-Pair Mechanisms", The Journal of Organic Chemistry, vol. 75, pp. 5164-5169, 2010. https://doi.org/10.1021/jo100920e
- J.R. Alvarez-Idaboy, L. Reyes, and J. Cruz, "A New Specific Mechanism for the Acid Catalysis of the Addition Step in the Baeyer−Villiger Rearrangement", Organic Letters, vol. 8, pp. 1763-1765, 2006. https://doi.org/10.1021/ol060261z
Tags: ATM, CF 3 CO, CH 3 CO, extraneous product, free energy, free energy barrier, recombination
Thanks for this, Henry! It is easy to forget the effect of the extruded acetic acid in the reaction. What about the radical pathway? It certainly occurs to some extent because the reaction produces a small amount of methane and carbon dioxide from decomposition of acetyl radicals.
Incidentally, the Boekelheide rearrangement is not obscure to us heterocyclic chemists!
Re radical pathway. Open shell systems are tricky to compare to closed shell ones on an even basis. One test that can be applied is the wavefunction stability to open shell unrestricted solutions. This shows that at the transition state for the closed shell, the wavefunction is indeed unstable to a triplet energy (doi 10.6084/m9.figshare.734289), but not by very much (5.2 kcal/mol). Such a value normally indicates a reaction only on the edge of being biradical (more typically for biradical like reactions, its closer to 10-12 kcal/mol. We have an article in press, 10.1021/01146k where a good example of this is shown). So yes, it may be that a small proportion of the reaction goes via biradicals.
From which one may gather that three pathways may indeed be accessible to this reaction, and that the exact balance between them may be influenced by solvation, concentration, perhaps even temperature.
It would be interesting to rerun the oxygen labelling experiments on this reaction in solvents of differing polarity. Perhaps the concerted pathway would then dominate in a non-polar medium. I don’t know if anyone has done this.