In the preceding post, I introduced Dewar’s π-complex theory for alkene-metal compounds, outlining the molecular orbital analysis he presented, in which the filled π-MO of the alkene donates into a Ag+ empty metal orbital and back-donation occurs from a filled metal orbital into the alkene π* MO. Here I play a little “what if” game with this scenario to see what one can learn from doing so.

Firstly, I will use Au+ instead of Ag+, so as to make a comparison with Pt2+ a little more direct. The electronic configurations are of course [Xe].4f14.5d10.6s0 and [Xe].4f14.5d8.6s0 respectively. I will also replace a simple ethene with cyclobutadiene, the intent here being that this cyclo-diene is a very much better π-donor due to its anti-aromatic character. It also now has the possibility of acting as a four or a two-electron donor. I started with M=Pt+[1] by adding another double bond to the structure of the ethene complex.
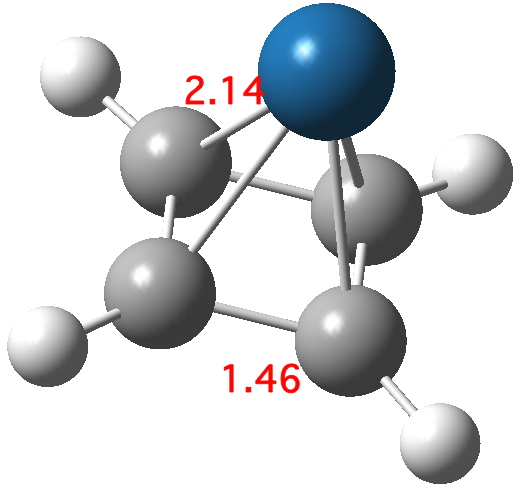
Optimising this starting structure in fact moves the metal and the final geometry has C4v symmetry; in other words the metal is bound symmetrically to all four carbons. The four C-C lengths are all the same (1.46Å) and strongly suggest that four electrons from the cyclobutadiene are participating in bonding; the Pt2+ is clearly capable of accepting four electrons, two into 6s0 and two into 5d8. In the process, the cyclobutadiene looses its antiaromaticity. The molecular orbitals of this species are all lovely; I illustrate just one below.
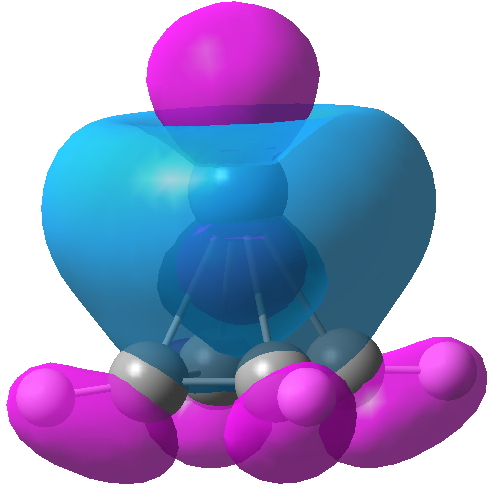
Click for 3D.
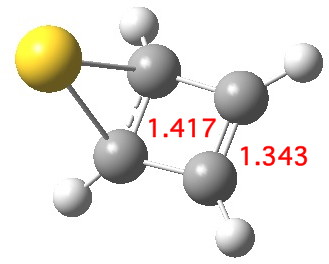
If the Pt in this C4v structure is mutated into Au+, the resulting optimised stationary point exhibits a negative force constant characteristic of a transition state[2]. As the d-shell is already fully, the Au can only accept two electrons, and this is therefore a nice illustration of the “18-electron” rule in operation. So, the Au+ complex must exist in at least one lower energy form. For example, one where the Au+ is coordinated to only one alkene is 94 kcal/mol lower in free energy.[3] This form results in electrons from the coordinated alkene being donated into the 6s Au orbital, and this action reduces the anti-aromaticity of the cyclobutadiene ring.
Another isomer also achieves this result, resulting in a further lowering in free energy of 11.0 kcal/mol[4] The anti-aromaticity this time is eliminated by forming an allyl cation on the ring. I have described this mode in another post, commenting on the effect when a guanidinium cation interacts with cyclobutadiene.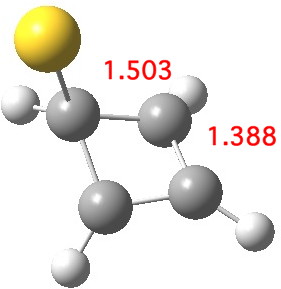
We have learnt that cyclobutadiene has many modes for eliminating 4n-electron antiaromaticity and other destabilising influences upon the ring. It can accept four electrons from a suitable acceptor (Pt2+), or two electrons from Au+ in two different ways.
Author
References
- H.S. Rzepa, "Gaussian Job Archive for C4H4Pt(2+)", 2013. https://doi.org/10.6084/m9.figshare.703546
- H.S. Rzepa, "Gaussian Job Archive for C4H4Au(1+)", 2013. https://doi.org/10.6084/m9.figshare.703547
- H.S. Rzepa, "Gaussian Job Archive for C4H4Au(1+)", 2013. https://doi.org/10.6084/m9.figshare.703576
- H.S. Rzepa, "Gaussian Job Archive for C4H4Au(1+)", 2013. https://doi.org/10.6084/m9.figshare.703577
Tags: African Union, alkene-metal compounds, empty metal orbital, energy, filled metal orbital, free energy, Historical, lower energy form, metal
[…] Other highly reactive species (cyclobutadiene is a well-known example) can often be tamed by trapping as a ligand coordinated to a metal and so one might speculate upon how C2 responds to the proximity […]