The previous post described how the acid catalysed ring opening of propene epoxide by an alcohol (methanol) is preceded by pre-protonation of the epoxide oxygen to form a “hidden intermediate” on the concerted intrinsic reaction pathway to ring opening. Here I take a look at the mechanism where a strong base is present, modelled by tetramethyl ammonium methoxide (R4N+.–OMe), for the two isomers R=Me; R’=Me, R”=H and R’=H, R”=Me.

As noted before[1], with alkoxide the dominant product is R’=Me, R”=H. A ωB97XD/6-311G(d,p)/SCRF=methanol) calculation of ΔΔG‡298 for the transition state for the process indeed favours this outcome, by 1.3 kcal/mol.[2],[3] This corresponds to ~90:10 for R’=Me, R”=H/R’=H, R”=Me. The barrier in this case is significantly smaller (~8 kcal/mol) than was observed for the route catalysed by trifluoracetic acid (~13 kcal/mol). From this emerges a possible explanation for the odd result I noted in the previous post; namely that the transition state ΔΔG‡ for the pure water catalysed reaction was 1.7 kcal/mol lower for the formation of 2-alkoxy-1-propanol than for 1-alkoxy-2-propanol (i.e. of R’=H, R”=Me vs R’=Me, R”=H), whereas experiment showed the dominant product was the latter. But the pure water bimolecular rate contains contributions not only from water acting as catalyst but also from the background [H+] and [HO–] rates as well (pKa methanol ~ 15.5, pKa water ~ 15.7). Of these three bimolecular rates, it is clear that the fastest is going to be the HO– catalysed one, and so it seems likely that the experimental result in pure water actually arises from catalysis by the [HO–] term and not by [H2O] itself.
The next task is to show how realistic the conventional “arrow pushing” for the reaction (shown above) actually is. Remember how, with acid catalysis, an IRC showed a proton transfer preceding the transition state. In contrast, the pure water mediated reaction showed no such pre-transfer, and instead revealed a hidden zwitterionic or ion-pair like intermediate occurring only AFTER the transition state. The MeO– catalysed reaction is similar, except of course that with the above models ion-pairs are present both at the start and end of the reaction, with the possibility of a third hidden ion pair occurring somewhere along the reaction pathway. The reactant ion pair of course does have to morph into the product ion pair by virtue of a proton transfer, and this occurs AFTER the transition state is passed, not before.
| R’=Me, R”=H | R’=H, R”=Me | |
| ΔΔG‡ | 0.0 | +1.3 |
|
IRC animation |
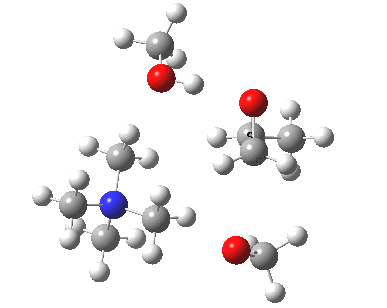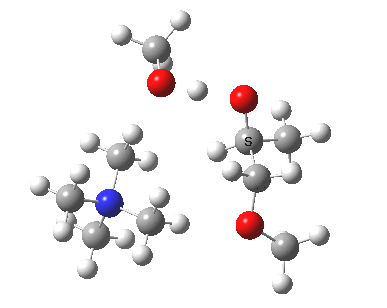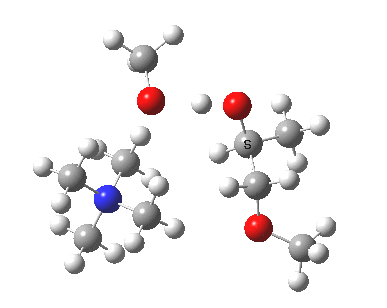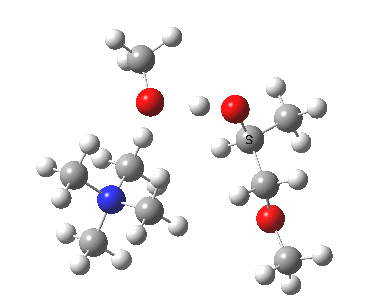 |
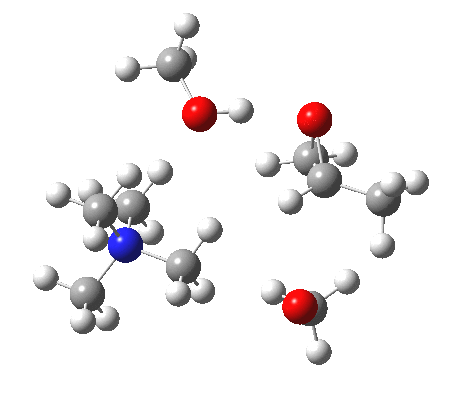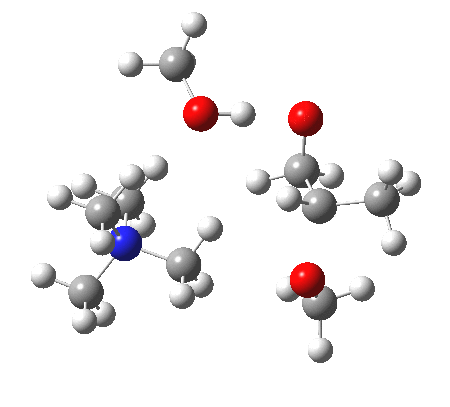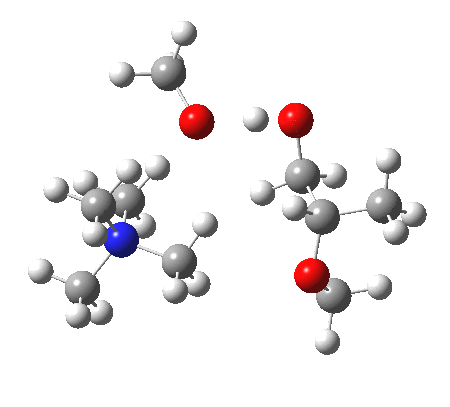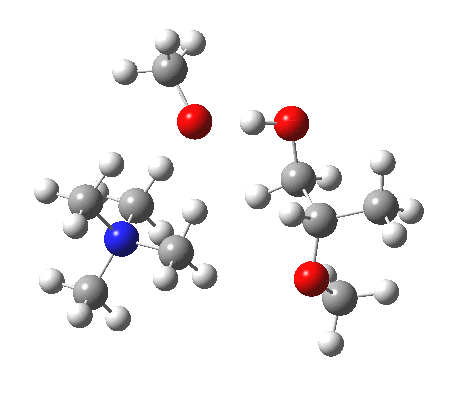 |
|
IRC energies |
 |
 |
|
IRC Grad |
 |
 |
| doi: | [4] | [5] |
The post-transition state proton transfer occurs driven by the need to minimise the charge separation in the ion-pair; a contact ion-pair is always likely to be more stable than a separated ion pair. For R’=H, R”=Me, a primary alkoxide is the initial product. This is relatively unstable, and so quite quickly (at about IRC 2.5) the system proceeds to acquire a proton from the adjacent methanol to reform a contact ion-pair. For R’=Me, R”=H, a secondary alkoxide is formed, which proves somewhat tardier in acquiring that proton, at IRC 9.5 in fact! Up to that point of course, the secondary alkoxide anion is a “hidden intermediate“, albeit very much on the verge of becoming a proper intermediate.
This third post on the topic I think ties up some of the loose ends, and seems to cast some interesting new light on what, at face value, seems a very simple organic reaction. There is, I think, still much to learn about such “simple” reactions.
Author
References
- H.C. Chitwood, and B.T. Freure, "The Reaction of Propylene Oxide with Alcohols", Journal of the American Chemical Society, vol. 68, pp. 680-683, 1946. https://doi.org/10.1021/ja01208a047
- H.S. Rzepa, "Gaussian Job Archive for C9H25NO3", 2013. https://doi.org/10.6084/m9.figshare.698066
- H.S. Rzepa, "Gaussian Job Archive for C9H25NO3", 2013. https://doi.org/10.6084/m9.figshare.698172
- H.S. Rzepa, "Gaussian Job Archive for C9H25NO3", 2013. https://doi.org/10.6084/m9.figshare.700641
- H.S. Rzepa, "Gaussian Job Archive for C9H25NO3", 2013. https://doi.org/10.6084/m9.figshare.700652