The recent release of the DataCite Data Citation corpus, which has the stated aim of providing “a trusted central aggregate of all data citations to further our understanding of data usage and advance meaningful data metrics” made me want to investigate what the current state of citing data in the area of chemistry might be. Chemistry is known to be a “data rich” science (as most of the physical sciences are) and here on this very blog I try to cite whenever possible the source(s) of the data that I often use when discussing a topic. Such citations are not necessarily the same as citing a journal source via e.g. its DOI, although of course one is very likely to find data associated with most articles nowadays, albeit almost entirely via any associated supporting information document. However the latter is often presented in a relatively unstructured (PDF) form, which does not adhere to what are called the “FAIR” guidelines of being findable, accessible, interoperable and reusable. Directly citing data is a way of improving its FAIR-characteristics. So what insights does the Data citation corpus reveal? Read the rest of this entry »
Data Citation – a snapshot of the chemical landscape.
February 26th, 2024Mechanistic templates computed for the Grubbs alkene-metathesis reaction.
February 19th, 2024Following on from my template exploration of the Wilkinson hydrogenation catalyst, I now repeat this for the Grubbs variant of the Alkene metathesis reaction. As with the Wilkinson, here I focus on the stereochemistry of the mechanism as first suggested by Chauvin[1], an aspect lacking in eg the Wikipedia entry. As before, the diagram below is hyperlinked to the appropriate data repository identifier so that you can go straight from the scheme to the data (Top level Data DOI: [2]).
References
- P. Jean‐Louis Hérisson, and Y. Chauvin, "Catalyse de transformation des oléfines par les complexes du tungstène. II. Télomérisation des oléfines cycliques en présence d'oléfines acycliques", Die Makromolekulare Chemie, vol. 141, pp. 161-176, 1971. https://doi.org/10.1002/macp.1971.021410112
- H. Rzepa, "Mechanistic templates computed for the Grubbs alkene-metathesis reaction.", 2024. https://doi.org/10.14469/hpc/13796
3D Molecular model visualisation: 3 Million atoms +
January 27th, 2024In the late 1980s, as I recollected here[1] the equipment needed for real time molecular visualisation as it became known as was still expensive, requiring custom systems such as Evans and Sutherland PS390 workstations. One major breakthrough in making such techniques generally available on less specialised equipment was achieved by Roger Sayle[2], then working at Imperial College around 1990 and using a Silicon Graphics workstation. He greatly optimised up the rendering algorithms by creating a program called RasMol (after his initials), which meant such visualisations could very rapidly also be achieved even on a personal computer. Moving from vector display technology (the PS390) to Raster/bitmap graphics had allowed spacefilling representations of molecules containing 100s if not 1000s of atoms – and in turn enabled the new World-Wide Web to exploit the technique.[3]
References
- H. Rzepa, "Computers 1967-2011: a personal perspective. Part 2. 1985-1989.", 2011. https://doi.org/10.59350/g4j62-4xk50
- R. Sayle, "RASMOL: biomolecular graphics for all", Trends in Biochemical Sciences, vol. 20, pp. 374-376, 1995. https://doi.org/10.1016/s0968-0004(00)89080-5
- H.S. Rzepa, B.J. Whitaker, and M.J. Winter, "Chemical applications of the World-Wide-Web system", Journal of the Chemical Society, Chemical Communications, pp. 1907, 1994. https://doi.org/10.1039/c39940001907
The Macintosh computer at 40.
January 25th, 2024On 24th January 1984, the Macintosh computer was released, as all the media are informing us. Apparently, some are still working. I thought I would give my own personal recollections of that period.
A mechanistic exploration of the Wilkinson hydrogenation catalyst. Part 1: Model templates
January 21st, 2024Geoffrey Wilkinson first reported his famous work on the hydrogenation catalyst that now bears his name in 1965[1] and I met him at Imperial College around 1969 and again when I returned there in 1977. He was still working on these catalysts then and I was privileged to collaborate with him on unravelling the NMR spectra of some of these compounds.[2],[3],[4]. During that period, computational modelling of the mechanisms of molecules containing transition elements was still in its infancy and I never extended my collaboration into this area at that time. Now, even if belatedly, I decided to explore this aspect and started to do this about two weeks ago. Here I thought that I would use this opportunity to show how I am going about it.
References
- J.F. Young, J.A. Osborn, F.H. Jardine, and G. Wilkinson, "Hydride intermediates in homogeneous hydrogenation reactions of olefins and acetylenes using rhodium catalysts", Chemical Communications (London), pp. 131, 1965. https://doi.org/10.1039/c19650000131
- K.W. Chiu, H.S. Rzepa, R.N. Sheppard, G. Wilkinson, and W. Wong, "Two-dimensional δ/J-resolved<sup>31</sup>P n.m.r. spectroscopy of [bis(diphenylphosphino)methane](trimethylphosphine)chlororhodium(<scp>I</scp>)", J. Chem. Soc., Chem. Commun., pp. 482-484, 1982. https://doi.org/10.1039/c39820000482
- C. Kwok W., C.G. Howard, H.S. Rzepa, R.N. Sheppard, G. Wilkinson, A.M. Galas, and M.B. Hursthouse, "Trimethyl and diethylphenylphosphine complexes of rhenium(I, III, IV, V) and their reactions. X-ray crystal structures of a bis(η5-cyclopentadienyl)-ethane-bridged dirhenium(I) complex obtained from phenylacetylene, tetrakis-(diethylphenylphosphine) (dinitrogen) hydridorhenium (I), tetrakis(trimethyl-phosphine) (η2-dimethylphosphinomethyl) rhenium(I) and tetrakis(trimethylphosphine) (iodo)methyl rhenium(III) iodide-tetramethylphosphonium iodide", Polyhedron, vol. 1, pp. 441-451, 1982. https://doi.org/10.1016/s0277-5387(00)86558-4
- K.W. Chiu, H.S. Rzepa, R.N. Sheppard, G. Wilkinson, and W. Wong, "Bis(diphenylphosphino)methane trimethylphosphine alkyl and η5-cyclopentadienyl compounds of rhodium(I); 31P{1H} two dimensional δ/J resolved and Overhauser effect nuclear magnetic resonance spectroscopy", Polyhedron, vol. 1, pp. 809-817, 1982. https://doi.org/10.1016/0277-5387(82)80008-9
Scholarly journals vs Scholarly Blogs.
January 12th, 2024First, a very brief history of scholarly publishing, starting in 1665[1] when scientific journals started to be published by learned societies. This model continued until the 1950s, when commercial publishers such as Pergamon Press started with their USP (unique selling point) of rapid time to publication of ~3 months,[2] compared to typical times for many learned society publishers of 2 years or longer. Fast forward another 50 years or so, and the commercial publishers were now dominating the scene, but the business model was still based on institutional subscriptions, whereby the institution rather than authors paid the costs of publication. As the number of journals expanded, even well-off institutions had to make difficult decisions on which subscriptions to keep and which to cancel. By the late 1990s the delivery model was changing from print to online, but the overall issue was that many scientists around the world no longer had access to many journals.
References
- H. Oldenburg, "Epistle dedicatory", Philosophical Transactions of the Royal Society of London, vol. 1, pp. i-ii, 1665. https://doi.org/10.1098/rstl.1665.0001
- D. Ginsburg, and W.J. Rosenfelder, "Alicyclic studies—X", Tetrahedron, vol. 1, pp. 3-8, 1957. https://doi.org/10.1016/0040-4020(57)85003-0
Macrocyclic peptide antibiotics – now Zosurabalpin – then antibacterial agents based on cyclic D,L-α-peptide architectures.
January 8th, 2024Zosurabalbin[1],[2], is receiving a great deal of attention as a new class of antibiotic which can target infections for which current treatment options are inadequate. It is a cyclic peptide and seeing this triggered memory of an earlier such species reported way back in 1995[3],[4]. This octa-peptide (YIJDIE, DOI: 10.5517/cc58gxs) was presumed to function in a novel manner, having linear water channels wide enough to form a molecular nanoscale pipe for a stream of water molecules to flow along. When inserted into the bacterial cell membrane via its lipophilic sidechains, it drained the bacterium of its cell water within seconds, thus killing it. A 3D model shows the effect very clearly.
References
- C. Zampaloni, P. Mattei, K. Bleicher, L. Winther, C. Thäte, C. Bucher, J. Adam, A. Alanine, K.E. Amrein, V. Baidin, C. Bieniossek, C. Bissantz, F. Boess, C. Cantrill, T. Clairfeuille, F. Dey, P. Di Giorgio, P. du Castel, D. Dylus, P. Dzygiel, A. Felici, F. García-Alcalde, A. Haldimann, M. Leipner, S. Leyn, S. Louvel, P. Misson, A. Osterman, K. Pahil, S. Rigo, A. Schäublin, S. Scharf, P. Schmitz, T. Stoll, A. Trauner, S. Zoffmann, D. Kahne, J.A.T. Young, M.A. Lobritz, and K.A. Bradley, "A novel antibiotic class targeting the lipopolysaccharide transporter", Nature, vol. 625, pp. 566-571, 2024. https://doi.org/10.1038/s41586-023-06873-0
- S. Hawser, N. Kothari, T. Valmont, S. Louvel, and C. Zampaloni, "2131. Activity of the Novel Antibiotic Zosurabalpin (RG6006) against Clinical <i>Acinetobacter</i> Isolates from China", Open Forum Infectious Diseases, vol. 10, 2023. https://doi.org/10.1093/ofid/ofad500.1754
- M.R. Ghadiri, K. Kobayashi, J.R. Granja, R.K. Chadha, and D.E. McRee, "The Structural and Thermodynamic Basis for the Formation of Self‐Assembled Peptide Nanotubes", Angewandte Chemie International Edition in English, vol. 34, pp. 93-95, 1995. https://doi.org/10.1002/anie.199500931
- S. Fernandez-Lopez, H. Kim, E.C. Choi, M. Delgado, J.R. Granja, A. Khasanov, K. Kraehenbuehl, G. Long, D.A. Weinberger, K.M. Wilcoxen, and M.R. Ghadiri, "Antibacterial agents based on the cyclic d,l-α-peptide architecture", Nature, vol. 412, pp. 452-455, 2001. https://doi.org/10.1038/35086601
Molecules of the year 2023 – part 2. A FAIR data comment on a Strontium Metallocene.
December 29th, 2023I will approach this example of a molecule-of-the-year candidate – in fact the eventual winner in the reader poll – from the point of view of data. Its a metallocene arranged in the form of a ring comprising 18 sub-units.[1] Big enough to deserve a 3D model rather than the static images you almost invariably get in journals (and C&EN). So how does one go to the journal and acquire the coordinates for such a model?
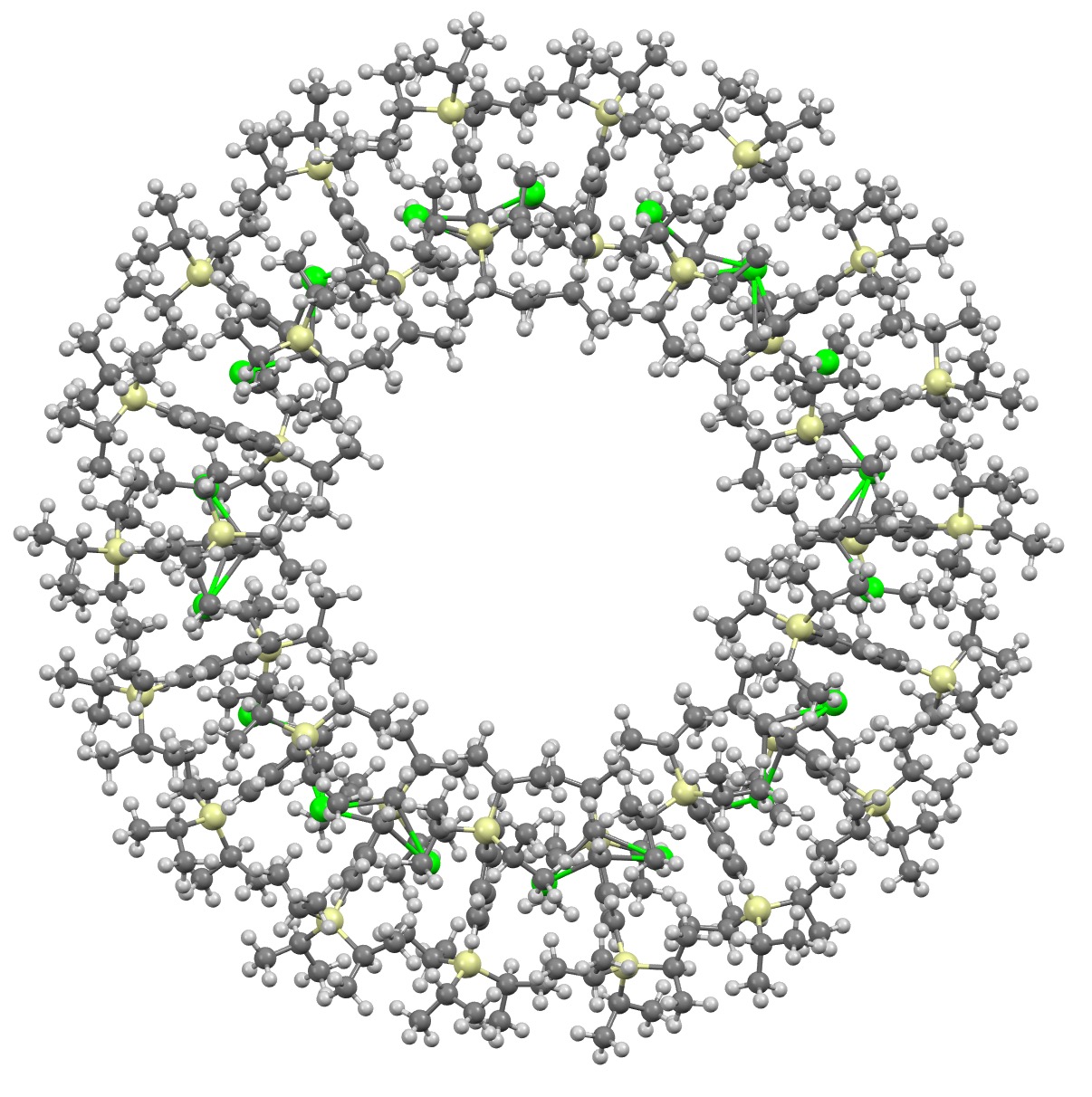
References
- L. Münzfeld, S. Gillhuber, A. Hauser, S. Lebedkin, P. Hädinger, N.D. Knöfel, C. Zovko, M.T. Gamer, F. Weigend, M.M. Kappes, and P.W. Roesky, "Synthesis and properties of cyclic sandwich compounds", Nature, vol. 620, pp. 92-96, 2023. https://doi.org/10.1038/s41586-023-06192-4
Molecules of the year: 2023
December 28th, 2023The Science education unit at the ACS publication C&EN publishes its list of molecules of the year (as selected by the editors and voted upon by the readers) in December. Here are some observations about three of this year’s batch. Read the rest of this entry »
The journey from Journal “ESI” to FAIR data objects: An eighteen year old (continuing) experiment.
December 10th, 2023Around 1996, journals started publishing what became known as “ESI” or electronic supporting information, alongside the articles themselves, as a mechanism for exposing the data associated with the research being reported and exploiting some of the new opportunities offered by the World Wide Web. From the outset, such ESI was expressed as a paginated Acrobat file, with the Web being merely a convenient document delivery mechanism. Such ESI would eventually reach more than 1000 such pages in length in some chemistry articles. The richer opportunities of Web interactivity were far less exploited. I have written about various aspects of this throughout this blog[1],[2],[3], together with one early compendium of our own data examples.[4] Here I update that compendium starting from 2005 to the current 2023 and add further information, being the current state of curation of some of these early examples. Curation became necessary because many of the earlier examples were no longer functional due to changes in the way journals expose these data objects or indeed changes at the data repository end of things over this 18 year period.
References
- H. Rzepa, "Four stages in the evolution of interactive ESI as part of articles in chemistry journals.", 2022. https://doi.org/10.59350/qypm4-qfv97
- H. Rzepa, "Web page decay and Journals: How an interactive "ESI" from 2006 was rescued.", 2022. https://doi.org/10.59350/cqesx-a0e83
- H. Rzepa, "Curating a nine year old journal FAIR data table.", 2017. https://doi.org/10.59350/z9g5j-r2p69
- H. Rzepa, "(Hyper)activating the chemistry journal.", 2009. https://doi.org/10.59350/wczky-8sf79