When methyl manganese pentacarbonyl is treated with carbon monoxide in e.g. di-n-butyl ether, acetyl manganese pentacarbonyl is formed. This classic experiment conducted by Cotton (of quadruple bond fame) and Calderazzo in 1962[1] dates from an era when chemists conducted extensive kinetic analyses to back up any mechanistic speculations. Their suggested transition state is outlined below. Here I subject their speculations to a quantum mechanical “reality check“.

The mechanism as above, formally at least is a pericyclic insertion of a carbene into a C-Mn bond. As the preamble to studying the mechanism, the following related reactions are of some interest:
- The addition of dichlorocarbene to ethene (see this blog for IRC)
- The addition of dichlorocarbene to butadiene (see this blog for IRC)
- The addition of carbon monoxide to ethene, for which the IRC is shown below

- The even simpler insertion of carbon monoxide to H2, for which the IRC is shown below

- And finally the insertion of carbon monoxide to methane, which is the closest analogy for us here.

- Notice that all of these reactions are asymmetric, and that the two new bonds forming to the carbon do so very asymmetrically/asynchronously, albeit in a concerted manner.
You might conclude from the above that extending this to identifying the transition state for insertion of carbon monoxide into a C-Mn bond is almost bound to reveal something interesting. Well, I was not able to locate (at the ωB97XD/6-311G(d,p)SCRF=di-n-butyl ether level) the carbon monoxide insertion transition state proposed by Cotton and Calderazzo (which of course does not mean it does not exist). But my efforts instead led to the following different course for the reaction (which is actually covered by the original authors with the statement “a mechanism involving a rapid pre-equilibrium to form MeCOMn(CO)4 cannot be ruled out at this stage“). Thus TS1 expresses that pre-equilibrium.

- The first stage involves the migration of the methyl group from Mn to an adjacent carbonyl group via TS1 to form Int1.

TS1. Click for 3D animation of transition mode.
- As the methyl group migrates to the adjacent carbonyl, it creates a putative vacant coordination site on the metal. This is re-occupied by the concerted formation of a C-H agostic interaction, exhibited by Int1. This new interaction now blocks any attempt by free carbon monoxide to insert into that erstwhile vacant site.
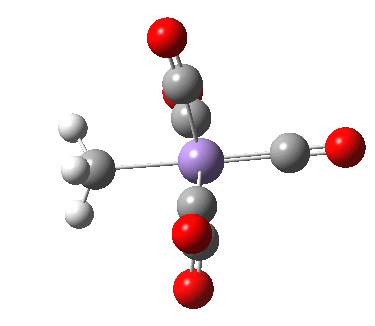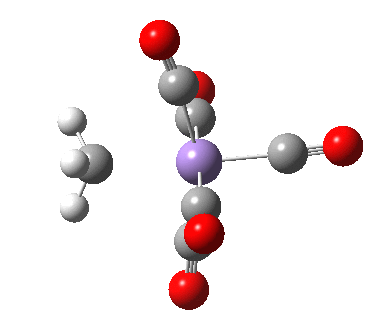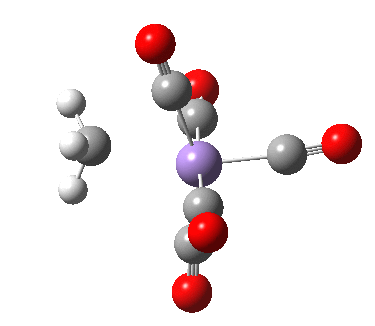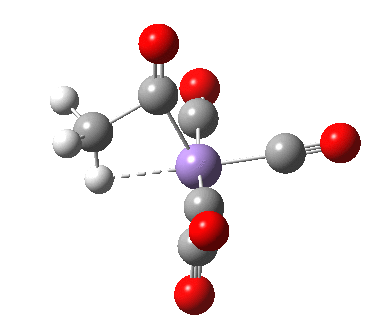

- Int1 now rearranges to form an isomer, Int2, in which the agostic C-H interaction is replaced by a Mn-π-interaction from the acetyl group. This creates trigonal bipyramidal coordination at the Mn. Int2 is 6.9 kcal/mol lower in free energy than Int1.

Int2. Click for 3D.
- A free carbon monoxide now attacks this intermediate via TS2 to coordinate onto the metal to reform octahedral coordination and complete the carbonylation reaction, and in the process converting the C=O π-complex into a Mn-C σ-complex.

TS2. Click for 3D.

- TS2, as with the transition state for alkene metathesis, reveals that bond formation between the incoming carbon of the carbon monoxide and the Mn has not yet started (3.05Å). Instead the barrier is largely induced by the need to reorganise the ligands present on the metal before new bonds can form.
- Which of TS1 (+ CO) or TS2 is higher in free energy? Using di-n-butyl ether as the simulated solvent, TS2 emerges as the higher by 4.2 kcal/mol.It is so in part because of the greater loss of entropy at the transition state for this latter geometry. In fact, the kinetics reported by Cotton and Calderazzo (ΔG‡303 20.6 kcal/mol, ΔS‡ -21.1 cal/mol) indicate the reaction is first order in [CO], which indeed implies that TS2 must be higher in energy (since TS1 does not depend on [CO]). The theory indicates ΔG‡298 16.4 kcal/mol, ΔS‡ -7.5 cal/mol for TS1 and ΔG‡298 20.6 kcal/mol, ΔS‡ -30.9 cal/mol for TS2. This level of agreement strongly supports the migration/addition pathway over the direct C-Mn insertion route.
I hope that by adding a layer of quantum mechanical interpretation to the original synthetic and kinetic study, we can learn a bit more about what sounds like a very simple reaction, but which has turned out to have wonderful and subtle twists and turns.
References
- F. Calderazzo, and F.A. Cotton, "Carbon Monoxide Insertion Reactions. I. The Carbonylation of Methyl Manganese Pentacarbonyl and Decarbonylation of Acetyl Manganese Pentacarbonyl", Inorganic Chemistry, vol. 1, pp. 30-36, 1962. http://dx.doi.org/10.1021/ic50001a008
Tags: agostic interaction, Interesting chemistry, pi complex, Reaction Mechanism
The following connection is an interesting one. The fixation of gaseous nitrogen by the nitrogenase enzyme is thought to proceed via a dihydrido Fe complex which undergoes reductive elimination of H2 gas to create a vacant coordination centre on a Fe atom. End-on coordination of a nitrogen molecule into this empty pocket is thus enabled. The nitrogen then goes on to receive two protons from adjacent S-H units and of course to ultimately become ammonia. CO is of course iso-electronic with N2.