In the previous post, I looked at the mechanism for 1,4-nucleophilic addition to an activated alkene (the Michael reaction). The model nucleophile was malonaldehyde after deprotonation and the model electrophile was acrolein (prop-2-enal), with the rate determining transition state being carbon-carbon bond formation between the two, accompanied by proton transfer to the oxygen of the acrolein.

Here I look at the effect of changing one of the aldehyde groups on the malonaldehyde to a variety of others and in particular how this might affect the relative timing of the C-C formation and the accompanying proton transfer to oxygen. Will this vary with substituents?
The activation free energies for TS2 are shown below, showing that as the acidity of the proton on the incipient nucleophile decreases along the series R=NO2 to R=H, the free energy barrier goes up.
| Substituent | ΔΔG298‡ (TS2) | Angle of approach |
|---|---|---|
|
NO2 |
11.5 | 110.8 |
|
CHO |
16.3 | 118.2 |
|
CN |
16.7 | 111.2 |
|
OMe |
31.9 | 121.8 |
|
H |
35.8 | 116.7 |
The asynchrony of the C-C formation and the PT is clearly shown for R=NO2. This can be seen most clearly when the gradient norm along the reaction path is plotted. This has TWO maxima at IRC 0.5 and 1.4, with a hidden (zwitterionic) intermediate in-between.
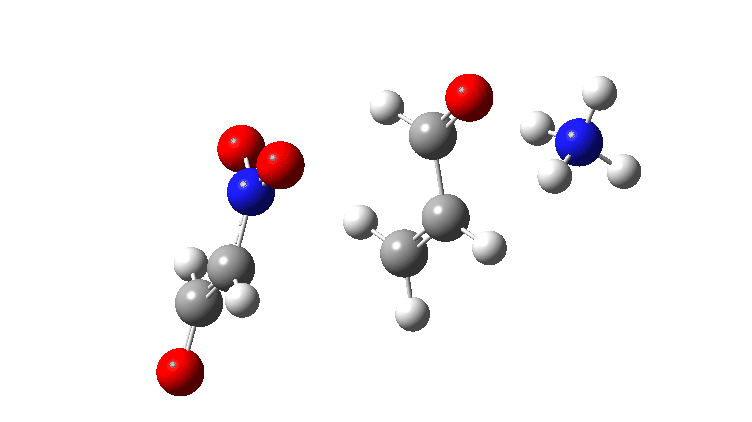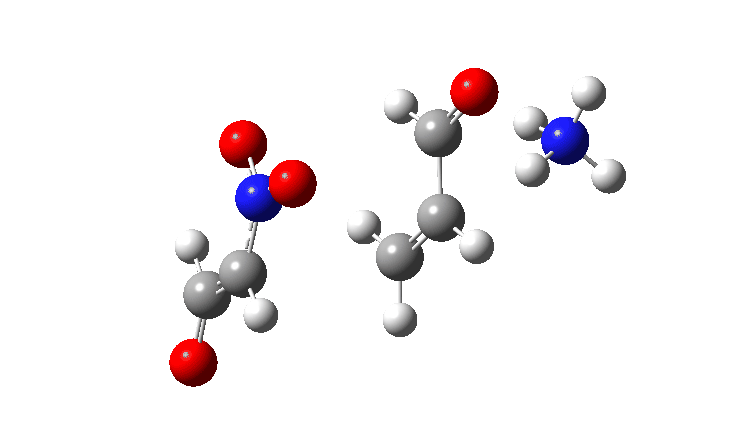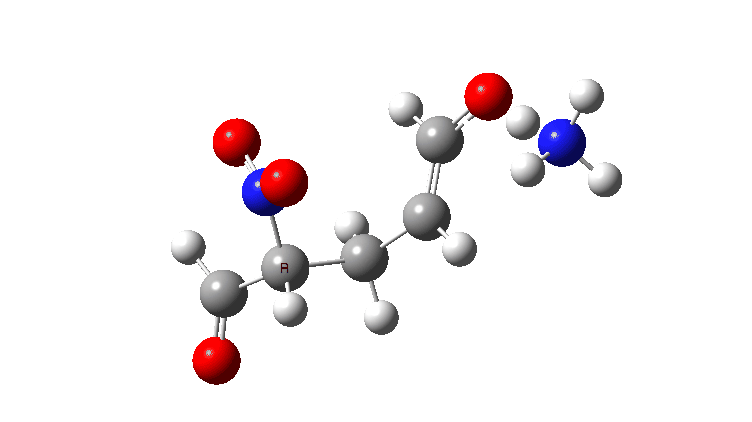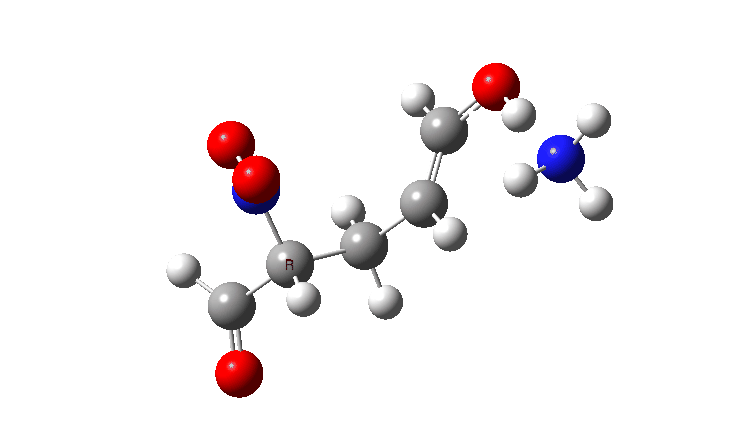

 For R=H the gradient norm peaks are at IRC 0.8 and 2.1; the reaction is equally asynchronous. If you are wondering why the barrier looks smaller for R=H than for R=NO2 it is because Int1 is a lot less stable for R=H (= more reactive) than for nitro.
For R=H the gradient norm peaks are at IRC 0.8 and 2.1; the reaction is equally asynchronous. If you are wondering why the barrier looks smaller for R=H than for R=NO2 it is because Int1 is a lot less stable for R=H (= more reactive) than for nitro. 

So this was a surprise in the end. Unlike substituent effects on electrophilic peracid epoxidation of an alkene,[1] nucleophilic addition to an alkene does not seem to exhibit a large substituent effect on its choreography.
References
- J.E.M.N. Klein, G. Knizia, and H.S. Rzepa, "Epoxidation of Alkenes by Peracids: From Textbook Mechanisms to a Quantum Mechanically Derived Curly‐Arrow Depiction", ChemistryOpen, vol. 8, pp. 1244-1250, 2019. http://dx.doi.org/10.1002/open.201900099