Bromoallene is a pretty simple molecule, with two non-equivalent double bonds. How might it react with an electrophile, say dimethyldioxirane (DMDO) to form an epoxide?[1] Here I explore the difference between two different and very simple approaches to predicting its reactivity. 
Both approaches rely on the properties of the reactant and use two types of molecule orbitals derived from its electronic wavefunction. The first of these is very well-known as the molecular orbital (MO), which has the property that it tends to delocalise over all the contributing atoms (the “molecule”). MOs are often used in this context; the highest energy occupied MO is thought of as being associated with the most nucleophilic (electron donating) regions of the molecule and so such a HOMO would be expected to predict the region of nucleophilic attack. The second is known as the natural bond orbital (NBO), which is evaluated in a manner that tends to localise it on bonds (the functional groups or reaction centres) and atom centres. What do these respective orbitals reveal for bromoallene?
| The MOs | |
|---|---|
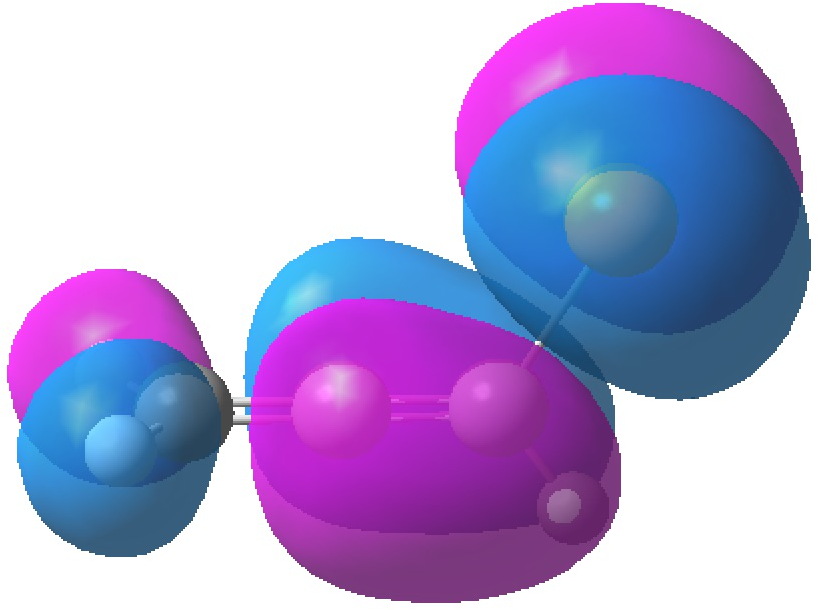
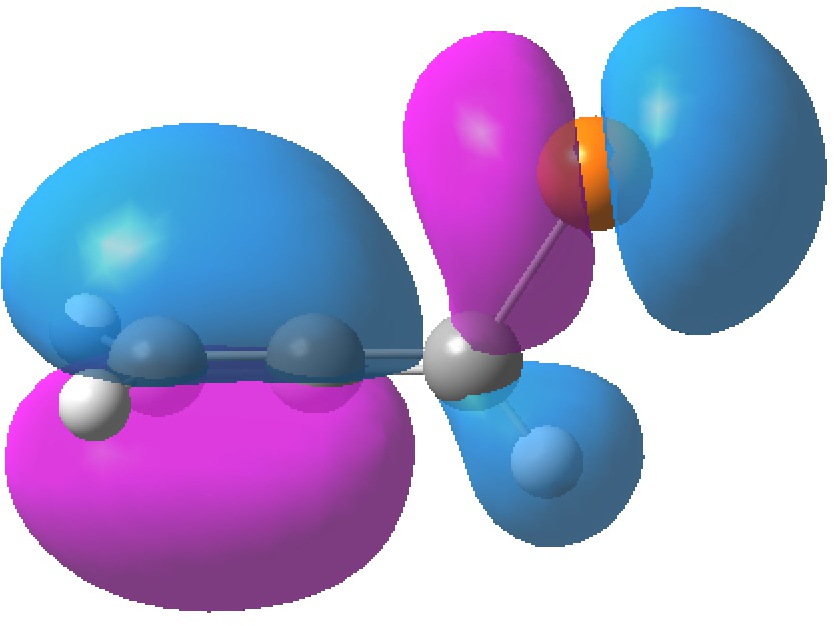
| HOMO, -0.3380 | HOMO-1, -0.3692 au |
 Click for 3D |
 Click for 3D |
| The NBOs | |
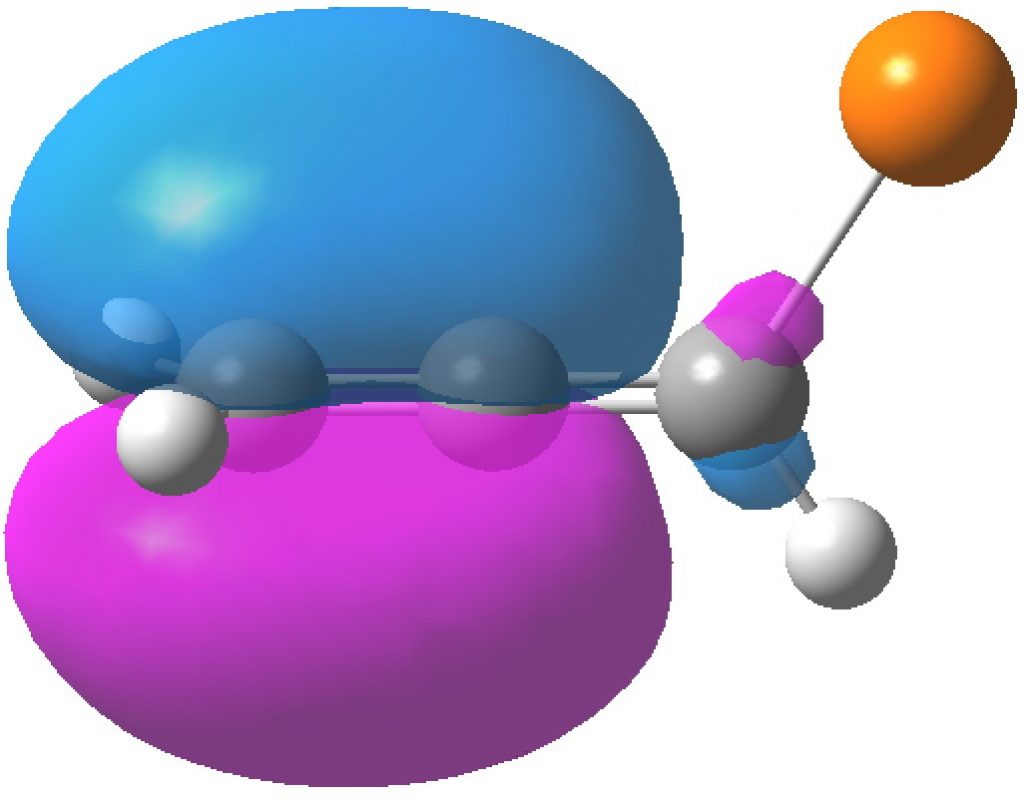
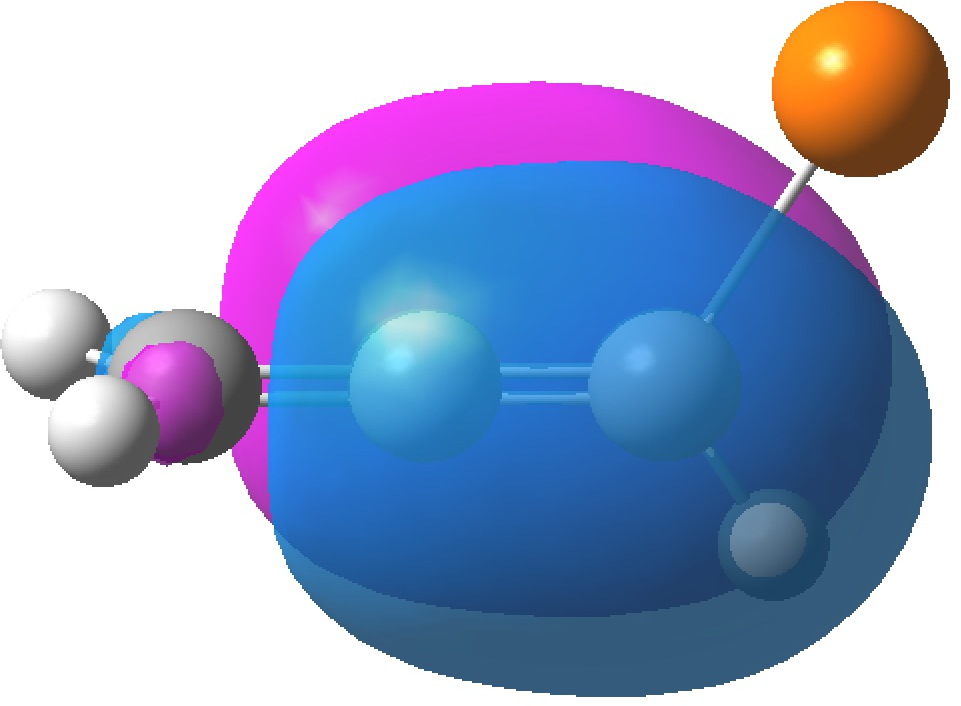
| HONBO, -0.3769 | HONBO-2, -0.3898 |
 Click for 3D |
 Click for 3D |
The table above shows the energies (in Hartrees) of the four relevant orbitals. The less negative (less stable) the orbital, the more nucleophilic it is. The (heavily) delocalized HOMO is located on the C=C bond bond carrying the C-Br bond, Δ1,2 alkene, but it also has a large component on the Br. The more stable HOMO-1 is located on the C=C bond located away from the Br, the Δ2,3 alkene and also with a (different type of) component on the Br.
In contrast, the HONBO is located on the Δ2,3 alkene and it is the HONBO-2 that is on the Δ1,2 alkene. Both these orbitals have very little “leakage” onto other atoms, they are almost completely localised.
Well, now we have a problem since these two analyses lead to diametrically opposing predictions! So what does experiment say? A recent article[1] addresses this issue by isolating the initially formed epoxide from reaction with DMDO and characterising it using crystallography. But here comes the catch; such isolation only proved possible if the allene was also substituted with large sterically bulky groups such as t-butyl or adamantyl. And the isolated product was the Δ1,2 epoxide. So does that mean that the MO method was correct and the NBO method wrong? Well, not necessarily. Those large groups play an additional role via steric effects. To factor in such effects one has to look at the transition state model for the reaction rather than depending purely on the reactant properties. And the steric effects in this case appear to win out over the electronic ones.[1]
The Klopman[2]-Salem[3] equation (shown in very simplified, and original, form below for just the covalent term) casts some light on what is going on. This term is a double summation over occupied/unoccupied (donor-acceptor) orbital interactions, involving the coefficients of the orbitals (the overlap integrals in effect) in the numerator and the energy difference between the occupied/unoccupied orbital pair as denominator.
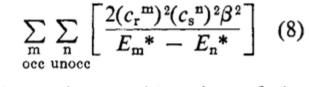
Performing such a double summation is rarely attempted; instead the equation is reduced to just one single term involving the donor of highest energy and the acceptor of lowest energy, ensuring the energy difference is a minimum and hence the term itself is (potentially) the largest in the summation. There is still the issue of the orbital coefficients, and here we get to the crux of the difference between the use of MOs and NBOs. You can see by inspection that the two π-MOs for bromoallene have different coefficients on the two atoms of interest, the two carbons of the double bond. One really has to evaluate the size of this term in the summation by using quantitative values for the respective coefficients and to very probably include the further terms in the summation for any other orbitals which also have significantly non-zero coefficients on these two atoms. But with the NBOs, the localisation procedure used to derive them has reduced the coefficients to just the carbon atoms and effectively no other atoms; all the other terms in the double summation in effect do drop out entirely. So with NBOs, the only number that matters is the energy difference between the occupied/empty orbitals (the denominator). But since the acceptor (the electrophile, DMDO in this case) is the same for both regiochemistries, things reduce even further to just comparing the donor energies for the two alternatives (Table above). The higher/less stable of these will have the greater contribution in the Klopman-Salem equation.
This little molecule teaches the important lesson that electronic and steric effects both play a role in directing reactions, and in this system they may well oppose each other. Simple interpretations based on reactant orbitals may give only a partial and even potentially misleading answer.
Author
References
- D. Christopher Braddock, A. Mahtey, H.S. Rzepa, and A.J.P. White, "Stable bromoallene oxides", Chemical Communications, vol. 52, pp. 11219-11222, 2016. https://doi.org/10.1039/c6cc06395k
- G. Klopman, "Chemical reactivity and the concept of charge- and frontier-controlled reactions", Journal of the American Chemical Society, vol. 90, pp. 223-234, 1968. https://doi.org/10.1021/ja01004a002
- L. Salem, "Intermolecular orbital theory of the interaction between conjugated systems. I. General theory", Journal of the American Chemical Society, vol. 90, pp. 543-552, 1968. https://doi.org/10.1021/ja01005a001
Tags: chemical bonding, chemical reaction, Chemistry, Delocalized electron, double bond, energy, energy difference, HOMO/LUMO, lowest energy, Molecular orbital, Natural bond orbital, Nature, Physics, Quantum chemistry, stable HOMO-1