Posts Tagged ‘higher energy’
Thursday, April 25th, 2019
Previously, I explored (computationally) the normal vibrational modes of Co(II)-tetraphenylporphyrin (CoTPP) as a “flattened” species on copper or gold surfaces for comparison with those recently imaged[1]. The initial intent was to estimate the “flattening” energy. There are six electronic possibilities for this molecule on a metal surface. Respectively positively, or negatively charged and a neutral species, each in either a low or a high-spin electronic state. I reported five of these earlier, finding each had quite high barriers for “flattening” the molecule. For the final 6th possibility, the triplet anion, the SCF (self-consistent-field) had failed to converge, but for which I can now report converged results.†
(more…)
References
- J. Lee, K.T. Crampton, N. Tallarida, and V.A. Apkarian, "Visualizing vibrational normal modes of a single molecule with atomically confined light", Nature, vol. 568, pp. 78-82, 2019. https://doi.org/10.1038/s41586-019-1059-9
Tags:019-1059-9, 10.1038, Biomolecules, Chelating agents, chemical bonding, Chemical compounds, Chemistry, Coordination chemistry, Coordination complex, Copper, copper metal surface, Cu–CO, E-type, energy, free energy, higher energy, impossible free energy, Inorganic chemistry, Jahn–Teller effect, lowest energy electronic state, Metabolism, metal, metal surface, modest planarisation energy, Molecule, Natural sciences, Physical sciences, planarisation, Porphyrin, reasonable energy, Resonance, Solid-state chemistry, sufficient energy, Teller, Tetraphenylporphyrin
Posted in Interesting chemistry | 1 Comment »
Monday, January 2nd, 2017
Here is an inside peek at another one of Derek Lowe’s 250 milestones in chemistry, the polymorphism of Ritonavir.[1] The story in a nutshell concerns one of a pharma company’s worst nightmares; a drug which has been successfully brought to market unexpectedly “changes” after a few years on market to a less effective form (or to use the drug term, formulation). This can happen via a phenomenon known as polymorphism, where the crystalline structure of a molecule can have more than one form.[2],[3],[4] In this case, form I was formulated into soluble tablets for oral intake. During later manufacturing, a new less-soluble form appeared and “within weeks this new polymorph began to appear throughout both the bulk drug and formulation areas“[1]
(more…)
References
- J. Bauer, S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter, and J. Morris, "Ritonavir: An Extraordinary Example of Conformational Polymorphism", Pharmaceutical Research, vol. 18, pp. 859-866, 2001. https://doi.org/10.1023/a:1011052932607
- J.D. Dunitz, and J. Bernstein, "Disappearing Polymorphs", Accounts of Chemical Research, vol. 28, pp. 193-200, 1995. https://doi.org/10.1021/ar00052a005
- D. Bučar, R.W. Lancaster, and J. Bernstein, "Disappearing Polymorphs Revisited", Angewandte Chemie International Edition, vol. 54, pp. 6972-6993, 2015. https://doi.org/10.1002/anie.201410356
- G.J.O. Beran, I.J. Sugden, C. Greenwell, D.H. Bowskill, C.C. Pantelides, and C.S. Adjiman, "How many more polymorphs of ROY remain undiscovered", Chemical Science, vol. 13, pp. 1288-1297, 2022. https://doi.org/10.1039/d1sc06074k
Tags:Carbamates, Chemistry, Derek Lowe, free energy, high energy process, High-energy rotations, higher energy, higher energy s-trans form, hydrogen bonding network, later manufacturing, Lipid polymorphism, low energy conformational effects, low energy rotations, lower energy rotation, Peek, Polymorphism, Protease inhibitors, Ritonavir, RTT, SN, Software engineering, Thiazoles, Ureas
Posted in Interesting chemistry | No Comments »
Tuesday, August 12th, 2014
One thing leads to another. Thus in the previous post, I described a thermal pericyclic reaction that appears to exhibit two transition states resulting in two different stereochemical outcomes. I noted that another such reaction appeared to be a [1,6] carousel migration in homotropylium cation,[1] where transition states for both retention and inversion of the configuration of the migrating group (respectively formally allowed and forbidden) were reported (scheme below). Here I explore this system further.  Firstly, the pathway leading to inversion.[2] The reaction path (ωB97XD/6-311G(d,p)/SCRF=chloroform) has got a very odd (table-top mountain) shape, whereby the region of the transition state (IRC = 0.0) is very flat, and the region close to reactant and (identical) product is very steep. The gradient norm shows this best, with sharp spikes at IRC ± 4.2. Something clearly is happening here to cause this behaviour. Before moving on to analyze this, I want you first to observe the methyl groups below. Note how one of them rotates at the start of the process, and the other at the end. I have elsewhere called this behaviour the methyl flag, and it is due to stereoelectronic re-alignments of the C-H groups accompanying the changes in the conjugated array.
Firstly, the pathway leading to inversion.[2] The reaction path (ωB97XD/6-311G(d,p)/SCRF=chloroform) has got a very odd (table-top mountain) shape, whereby the region of the transition state (IRC = 0.0) is very flat, and the region close to reactant and (identical) product is very steep. The gradient norm shows this best, with sharp spikes at IRC ± 4.2. Something clearly is happening here to cause this behaviour. Before moving on to analyze this, I want you first to observe the methyl groups below. Note how one of them rotates at the start of the process, and the other at the end. I have elsewhere called this behaviour the methyl flag, and it is due to stereoelectronic re-alignments of the C-H groups accompanying the changes in the conjugated array. 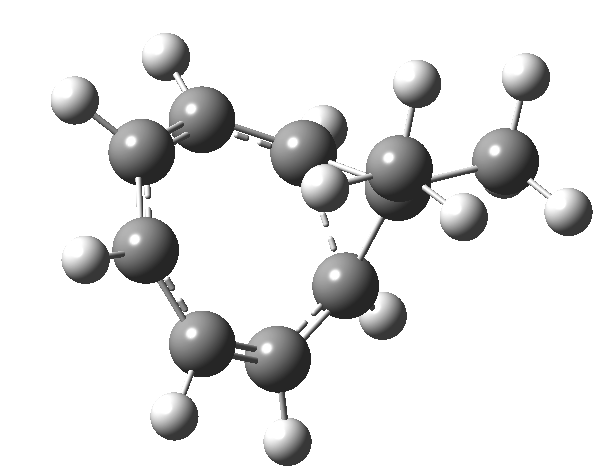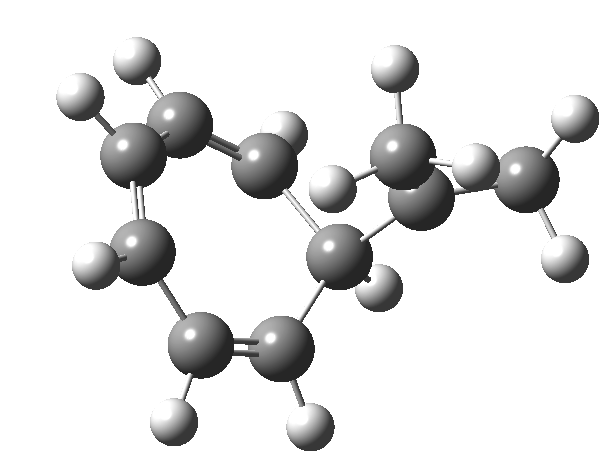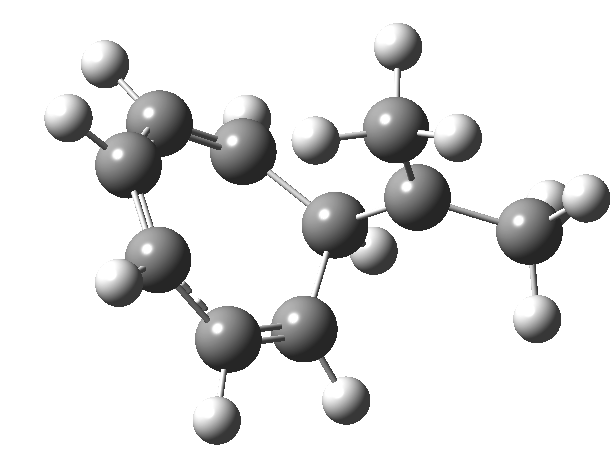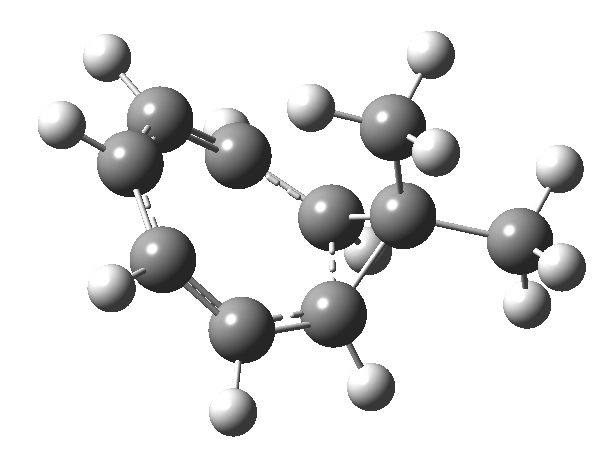

 The homotropylium cation is said to be homoaromatic, indicating that cyclic conjugation can be maintained across a ring in which the σ framework is interrupted at one point. A NICS probe placed at the ring critical point of this molecule reveals a chemical shift of -11.3 ppm[3], very similar to eg that obtained for benzene itself. The three highest doubly occupied NBOs (below) show two normal π-type orbitals and one rather different one that spans the homo-bond (the MOs, before you ask, are a bit of a mess, with lots of mixed contributions from other parts of the σ framework).
The homotropylium cation is said to be homoaromatic, indicating that cyclic conjugation can be maintained across a ring in which the σ framework is interrupted at one point. A NICS probe placed at the ring critical point of this molecule reveals a chemical shift of -11.3 ppm[3], very similar to eg that obtained for benzene itself. The three highest doubly occupied NBOs (below) show two normal π-type orbitals and one rather different one that spans the homo-bond (the MOs, before you ask, are a bit of a mess, with lots of mixed contributions from other parts of the σ framework).
(more…)
References
- A.M. Genaev, G.E. Sal’nikov, and V.G. Shubin, "Energy barriers to carousel rearrangements of carbocations: Quantum-chemical calculations vs. experiment", Russian Journal of Organic Chemistry, vol. 43, pp. 1134-1138, 2007. https://doi.org/10.1134/s1070428007080076
- H.S. Rzepa, "Gaussian Job Archive for C10H13(1+)", 2014. https://doi.org/10.6084/m9.figshare.1134556
- H.S. Rzepa, "Gaussian Job Archive for C10H13(1+)", 2014. https://doi.org/10.6084/m9.figshare.1135694
Tags:chemical shift, higher energy, Sangean Table Top Portable Audio Device
Posted in pericyclic, reaction mechanism | 5 Comments »
Sunday, January 6th, 2013
The benzidine rearrangement is claimed to be an example of the quite rare [5,5] sigmatropic migration[1], which is a ten-electron homologation of the very common [3,3] sigmatropic reaction (e.g. the Cope or Claisen). Some benzidine rearrangements are indeed thought to go through the [3,3] route[2]. The topic has been reviewed here[3].
(more…)
References
- H.J. Shine, K.H. Park, M.L. Brownawell, and J. San Filippo, "Benzidine rearrangements. 19. The concerted nature of the one-proton rearrangement of 2,2'-dimethoxyhydrazobenzene", Journal of the American Chemical Society, vol. 106, pp. 7077-7082, 1984. https://doi.org/10.1021/ja00335a035
- H.J. Shine, L. Kupczyk-Subotkowska, and W. Subotkowski, "Heavy-atom kinetic isotope effects in the acid-catalyzed rearrangement of N-2-naphthyl-N'-phenylhydrazine. Rearrangement is shown to be a concerted process", Journal of the American Chemical Society, vol. 107, pp. 6674-6678, 1985. https://doi.org/10.1021/ja00309a041
- H.J. Shine, "Reflections on the π‐complex theory of benzidine rearrangements", Journal of Physical Organic Chemistry, vol. 2, pp. 491-506, 1989. https://doi.org/10.1002/poc.610020702
Tags:higher energy, Historical, π-complex, pericyclic, Reaction Mechanism
Posted in Interesting chemistry | 4 Comments »
Friday, January 21st, 2011
Much of chemistry is about bonds, but sometimes it can also be about anti-bonds. It is also true that the simplest of molecules can have quite subtle properties. Thus most undergraduate courses in chemistry deal with how to describe the bonding in the diatomics of the first row of the periodic table. Often, only the series C2 to F2 is covered, so as to take into account the paramagnetism of dioxygen, and the triple bonded nature of dinitrogen (but never mentioning the strongest bond in the universe!). Rarely is diberyllium mentioned, and yet by its strangeness, it can also teach us a lot of chemistry.
(more…)
Tags:diberyllium, energy, excited state, higher energy, higher energy state, pence, Tutorial material
Posted in Interesting chemistry | 6 Comments »
Saturday, June 6th, 2009
The iconic diagram below represents a cornerstone of organic chemistry. Generations of chemists have learnt early on in their studies of the subject that these two representations of where the electron pairs in benzene might be located (formally called electronic resonance or valence bond forms) each contribute ~50% to the overall wavefunction, and that the real electronic description is in effect an average of these two (that is the implied meaning of the double headed arrow). This means that the six C-C bonds in benzene must all be of equal length. The diagrams, everyone knows, do not mean that benzene has three short and three long C-C bonds.
(more…)
Tags:energy, energy levels, higher energy, Interesting chemistry, π-electron resonance energy, π-energy, π-resonance energy, pseudo Jahn-Teller, resonance energy, so-called energy degenerate orbital
Posted in Interesting chemistry | 16 Comments »