In the previous posts, I explored reactions which can be flipped between two potential (stereochemical) outcomes. This triggered a memory from Alex, who pointed out this article from 1999[1] in which the nitrosonium cation as an electrophile can have two outcomes A or B when interacting with the electron-rich 2,3-dimethyl-2-butene.  NMR evidence clearly pointed to the π-complex A as being formed, and not the cyclic nitrosonium species B (X=Al4–). If you are wondering where you have seen an analogy for the latter, it would be the species formed when bromine reacts with an alkene (≡ Br+, X=Br– or Br3–). The two structures are shown below[1]
NMR evidence clearly pointed to the π-complex A as being formed, and not the cyclic nitrosonium species B (X=Al4–). If you are wondering where you have seen an analogy for the latter, it would be the species formed when bromine reacts with an alkene (≡ Br+, X=Br– or Br3–). The two structures are shown below[1] 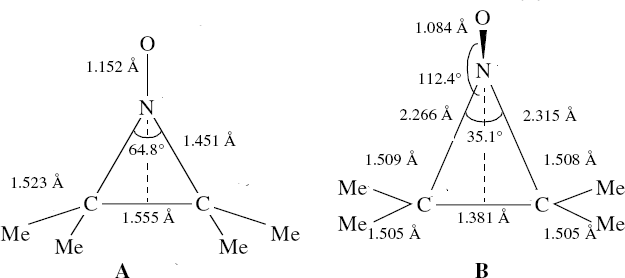 Since the topic that sparked this concerned pericyclic reactions, it seemed possible that if it had been formed, species B would immediately undergo a pericyclic electrocyclic reaction to form the rather odd-looking cation C, which might then be trapped by eg X(-) to form the nitrone D. So this post is an exploration of what happens when X-NO (X= CF3COO, trifluoracetate) interacts with 2,3-dimethyl-2-butene, as an illustration of what can be achieved nowadays from about 2 days worth of dry-lab computation as a prelude to e.g. an experiment in the wet-lab (it would take a little more than two days to achieve the latter I suspect). Hence computationally directed synthesis. The model is set up as ωB97XD/6-311G(d,p)/SCRF=chloroform. A transition state is located[2] and the resulting IRC (below) [3] does not quite have the outcome the above scheme would suggest.
Since the topic that sparked this concerned pericyclic reactions, it seemed possible that if it had been formed, species B would immediately undergo a pericyclic electrocyclic reaction to form the rather odd-looking cation C, which might then be trapped by eg X(-) to form the nitrone D. So this post is an exploration of what happens when X-NO (X= CF3COO, trifluoracetate) interacts with 2,3-dimethyl-2-butene, as an illustration of what can be achieved nowadays from about 2 days worth of dry-lab computation as a prelude to e.g. an experiment in the wet-lab (it would take a little more than two days to achieve the latter I suspect). Hence computationally directed synthesis. The model is set up as ωB97XD/6-311G(d,p)/SCRF=chloroform. A transition state is located[2] and the resulting IRC (below) [3] does not quite have the outcome the above scheme would suggest. 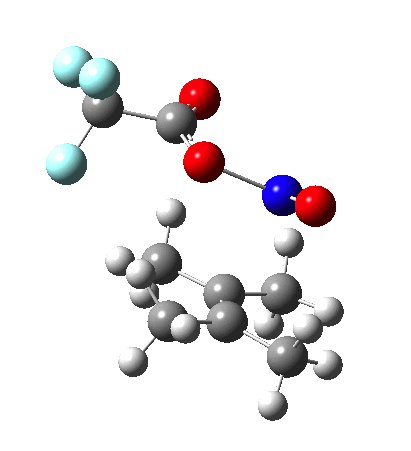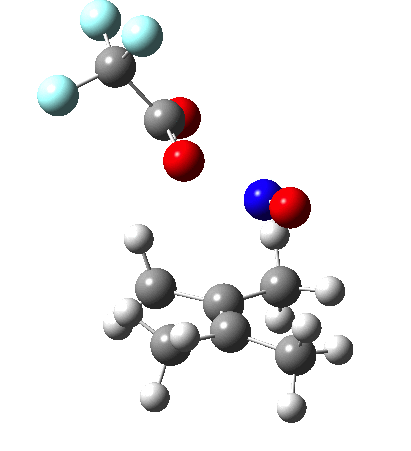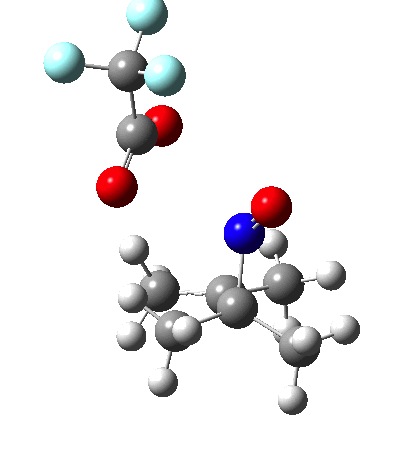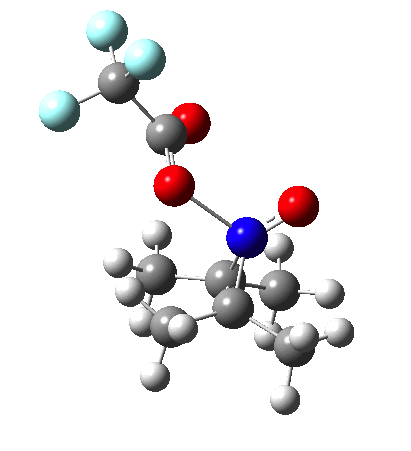

 Neither A nor B is formed; instead it is the tetrahedral species E, which is ~15 kcal/mol endothermic.
Neither A nor B is formed; instead it is the tetrahedral species E, which is ~15 kcal/mol endothermic.  I should immediately point out that this is not inconsistent with the formation of A as previously characterised[1]. That is because this experiment was conducted with a non-nucleophilic counter-anion (X=Al4–), whereas in the computational simulation above, we have a nucleophilic anion (X= CF3CO2–). What a difference the inclusion of a counter-ion in the calculation can have! The barrier however (~35 kcal/mol) is a little too high for a facile thermal reaction. In the second of this two-stage reaction, E now ring-opens to form the anticipated D[4] with quite a small barrier of ~6 kcal/mol, but a highly exothermic outcome. I ask this question about it; can this still be described as a pericyclic process? (there is some analogy to the electrocyclic ring opening of a cyclopropyl tosylate).
I should immediately point out that this is not inconsistent with the formation of A as previously characterised[1]. That is because this experiment was conducted with a non-nucleophilic counter-anion (X=Al4–), whereas in the computational simulation above, we have a nucleophilic anion (X= CF3CO2–). What a difference the inclusion of a counter-ion in the calculation can have! The barrier however (~35 kcal/mol) is a little too high for a facile thermal reaction. In the second of this two-stage reaction, E now ring-opens to form the anticipated D[4] with quite a small barrier of ~6 kcal/mol, but a highly exothermic outcome. I ask this question about it; can this still be described as a pericyclic process? (there is some analogy to the electrocyclic ring opening of a cyclopropyl tosylate). 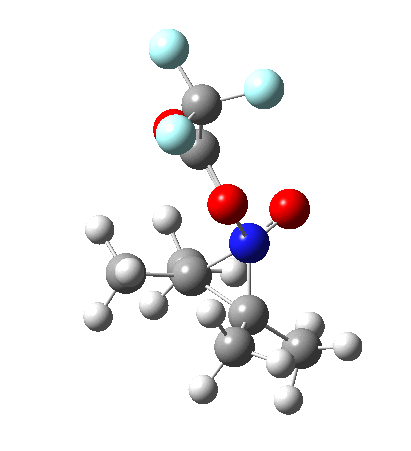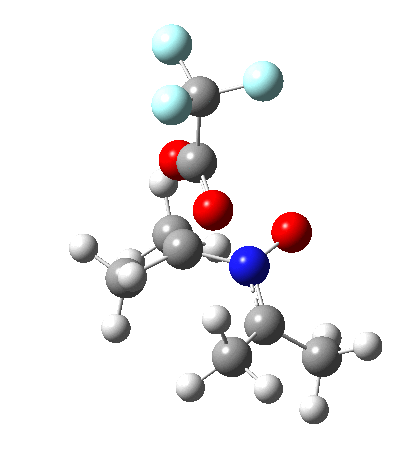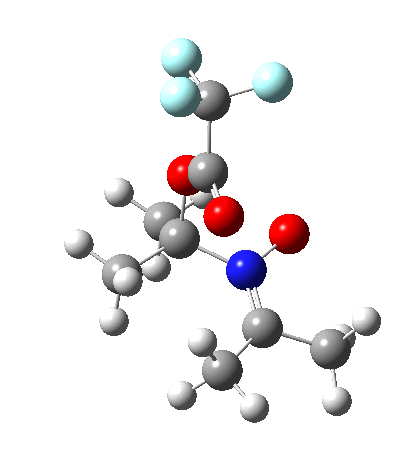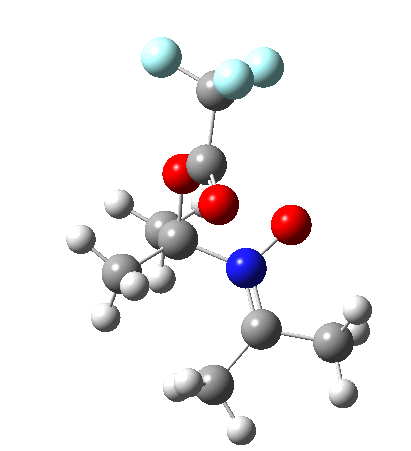
 So what are the conclusions? Well, because of the rather high initial barrier, the alkene will need activation (by electron donating substituents, perhaps OMe) for the reaction to become more viable. But if it works, it could be an interesting synthesis of nitrones (I have not yet searched to find out if the reaction is actually known).
So what are the conclusions? Well, because of the rather high initial barrier, the alkene will need activation (by electron donating substituents, perhaps OMe) for the reaction to become more viable. But if it works, it could be an interesting synthesis of nitrones (I have not yet searched to find out if the reaction is actually known).
References
- G.I. Borodkin, I.R. Elanov, A.M. Genaev, M.M. Shakirov, and V.G. Shubin, "Interaction in olefin–NO+ complexes: structure and dynamics of the NO+–2,3-dimethyl-2-butene complex", Mendeleev Communications, vol. 9, pp. 83-84, 1999. https://doi.org/10.1070/mc1999v009n02abeh000995
- H.S. Rzepa, "C8H12F3NO3", 2014. https://doi.org/10.14469/ch/24979
- H.S. Rzepa, "Gaussian Job Archive for C8H12F3NO3", 2014. https://doi.org/10.6084/m9.figshare.1162797
- H.S. Rzepa, "Gaussian Job Archive for C8H12F3NO3", 2014. https://doi.org/10.6084/m9.figshare.1162676
