Birch reduction of benzene itself results in 1,4-cyclohexadiene rather than the more stable (conjugated) 1,3-cyclohexadiene. Why is this?

The mechanism, as elaborated in the previous two posts, involves a one-electron transfer from a sodium atom to form the radical anion, which is then protonated in a second step, and this is again reduced to form a pentadienyl anion in the penultimate step.[1] The question now becomes why does this anion protonate to give predominantly the less stable diene product? The answer involves the actual structure of this anion. A calculation at the ωB97XD/6-311+G(d,p)/SCRF=acetonitrile level for the ion pair comprising the cyclohexadienyl anion and a Na(NH3)3+ counterion is shown below.

Structure of the cyclohexadienyl ion pair. Click for 3D.
From this, it appears that the sodium cation is η2 coordinated to each of two relatively localised double bonds (1.37Å), resulting in the negative charge accumulating on just the one carbon (red arrow), this being the carbon that then exclusively receives a final proton. The highest energy (-0.115 au) natural bond orbital (NBO) also emerges as being located on this carbon (the next two highest energy NBOs only come in at -0.303 au, and reside on each of the localised alkene bonds).

The highest energy NBO orbital. Click for 3D.
The molecular electrostatic potential in effect integrates over all the electrons (not just those in the highest orbital), resulting in a function that measures the attractiveness of any point to a proton (red). It too shows that the most attractive region (red) for a proton is again on this carbon.

Molecular electrostatic potential. Click for 3D.
There is even evidence from crystal structures that this sort of motif is possible. Thus the dianion of 1,4-diphenylbenzene (with two Na(thf)3+ counter-ions) reveals[2] this type of coordination. The buckling seen in the above mono-anion is inhibited by the presence of cations on both sides of the di-anion, but the pattern of short/long bonds seen above also manifests in the crystal structure.

Crystal model. Click for 3D.
So the take home message is that the counter-ion (solvated sodium cations) in the Birch reduction of benzene itself may coordinate to the anionic intermediates in the reductive process, and the resulting geometry of this ion-pair determines the eventual product of protonation.
References
- H.E. Zimmerman, and P.A. Wang, "Regioselectivity of the Birch reduction", Journal of the American Chemical Society, vol. 112, pp. 1280-1281, 1990. http://dx.doi.org/10.1021/ja00159a078
- J.H. Noordik, H.M. Doesburg, and P.A.J. Prick, "Structures of the sodium–p-terphenyl ion pairs: disodium terphenylide–tetrahydrofuran (1/6) and disodium diterphenylide terphenyl–1,2-dimethoxyethane (1/6)", Acta Crystallographica Section B Structural Crystallography and Crystal Chemistry, vol. 37, pp. 1659-1663, 1981. http://dx.doi.org/10.1107/S0567740881006833
Tags: Birch reduction, energy, eventual product, less stable diene product, Reaction Mechanism, Tutorial material
Someone asked me the question “can a molecular orbital (MO) be used instead of the fancy NBO in the post?”. Well, here it is:
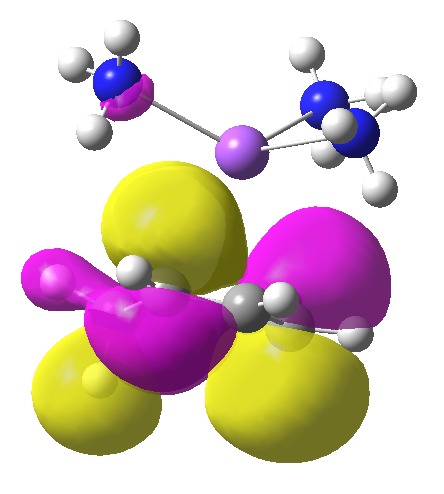
As befits a molecular orbital, it is heavily delocalised around the six-membered ring (including C-H bonds, which do not seem entirely pertinent to the problem being discussed). Quite possibly the coefficients of this orbital on the carbon atom which ends up being protonated are larger than any other position, but the effect is a nuance rather than a “in-your-face” one.
Things get a bit more tricky if one poses the question “when should I use a localised (NBO) rather than a molecular orbital”? I do not suppose there is any simple answer to that question. I would comment that when one is dealing with properties which truly are associated with the whole molecule rather than just a single bond, MOs come into their own. One such property for example is aromaticity.
Finally, I would note that bonds themselves can be characterised by not only orbitals, but various other methods. These include quantum topology (also called QTAIM), which looks at the topological properties of the electron density, and entities known as “disynaptic basins in the electron localisation function”.
[…] Henry Rzepa Chemistry with a twist « The mechanism of the Birch reduction. Part 3: reduction of benzene […]
Dear Henry, Your opening statement is a bit misleading since CCC-BDB/NIST data documents the only 0.67 kcal/mol greater stability of 1,3- over 1,4-cyclohexadiene. I teach my students that this small difference is due to the 4-fold vs. 2-fold hyperconjugation advantage of the non-conjugated 1,4-isomer (along with the lower strain of its planar D2h conformation). The latter has optimum torsional arrangements and less angle strain. Note that the model CCC angles are actually 126° in propene and 112° in propane (not 120°!).
Dear Paul,
Well, even a .670 kcal (I presume this is free energy and not total energy?) difference would result (at 270K) in a ratio of 3.5:1 in favour of the 1,3-diene over 1,4-diene if it were an equilibrium process. I have not looked up what the actual difference is, but presume it is <1:1. This would still need an explanation over and above the hyperconjugation of product etc etc.