A recent article reports, amongst other topics, a computationally modelled reaction involving the capture of molecular hydrogen using a substituted borane (X=N, Y=C).[cite]10.1073/pnas.1709586114[/cite] The mechanism involves an initial equilibrium between React and Int1, followed by capture of the hydrogen by Int1 to form a 5-coordinate borane intermediate (Int2 below, as per Figure 11).‡ This was followed by assistance from a proximate basic nitrogen to complete the hydrogen capture via a TS involving H-H cleavage. The forward free energy barrier to capture was ~11 kcal/mol and ~4 kcal/mol in the reverse direction (relative to the species labelled Int1), both suitably low for reversible hydrogen capture. Here I explore a simple variation to this fascinating reaction.∞

This variation involves transposing X and Y such that Y=N+ and X=C– to form a carbon ylide such that X=C becomes much more nucleophilic than the original nitrogen nucleophile. An animation of the full IRC† (intrinsic reaction coordinate computed at ωB97XD/cc-pvtz; FAIR data doi: 10.14469/hpc/2704) is shown below.
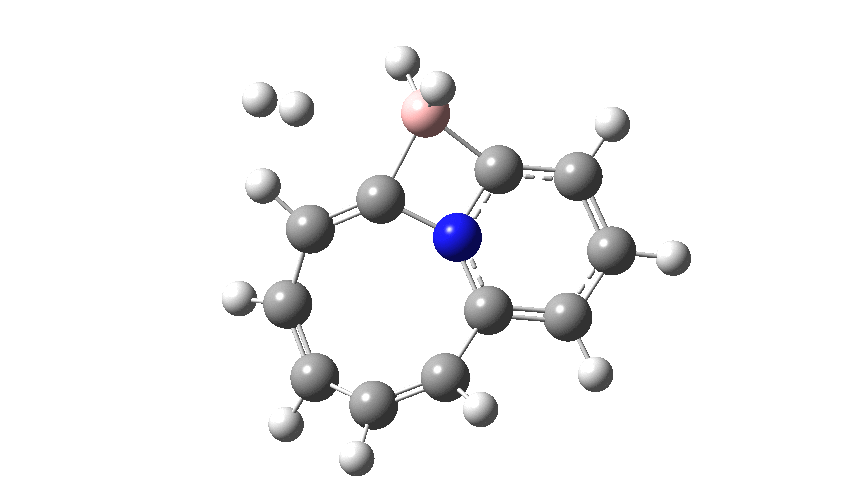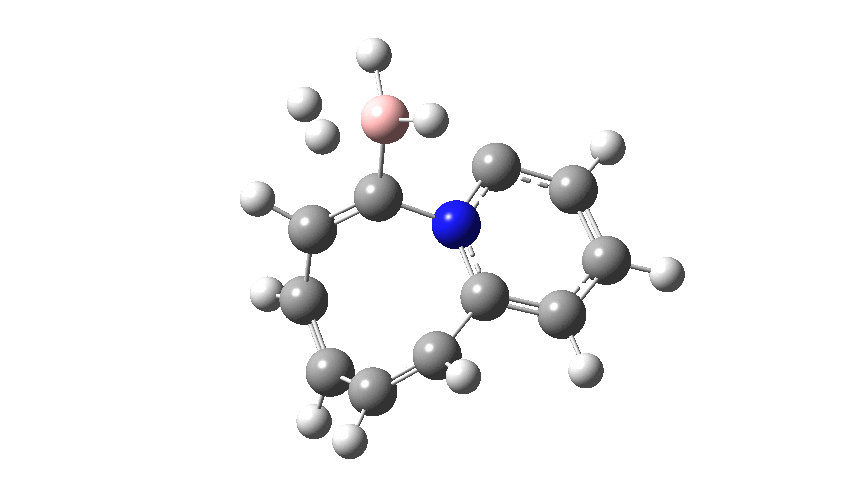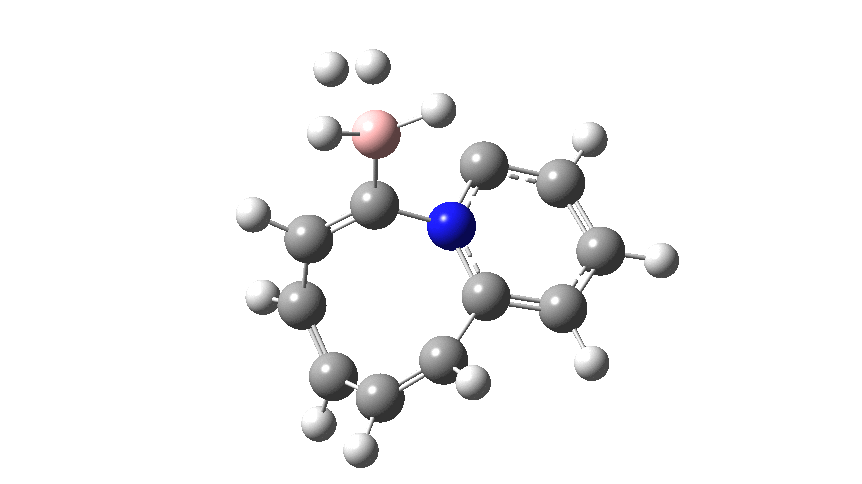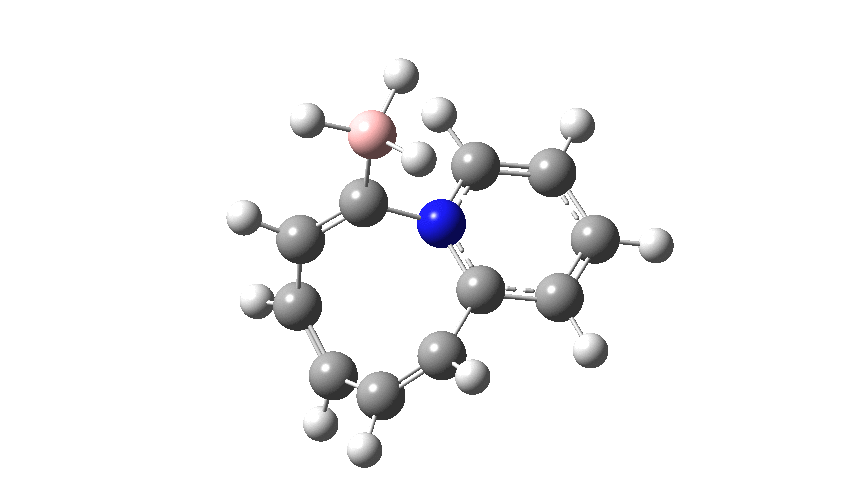
The profile shows that the reaction is concerted between the species labelled React and Prod; no sign of Int1 and Int2!
- The region IRC -12 to -5 involves B-C bond cleavage. Because the C is so very nucleophilic, the 4-ring species labelled React becomes very stable and opening it requires a high barrier.
- Between IRC -5 and 0, the BH2 group rotates, losing its original interaction with the C to slowly create an empty acceptor orbital on the boron which can then interact with the incoming hydrogen.
- At IRC= 0 (the transition state) the hydrogen has been captured by the boron to form a 5-coordinate species, in a manoeuvre that reminds one of the orbital capture of satellites by planets on their way to the outer reaches of the solar system. If the barrier to this capture is computed from IRC= -4 (the region of Int2) it is very much lower than the original system[cite]10.1073/pnas.1709586114[/cite], again a reflection of the higher nucleophilicity of X=C–.
- The fly past continues until IRC= +7, at which point one end of the bound hydrogen has become suitably orientated to interact with the nucleophilic carbon via lone-pair donation into the acceptor H-H σ* orbital, thus helping to break it.
- By IRC= +9, the H-H cleavage is complete.
- By IRC= +13 the reaction has reached Prod, being overall ~ -12 kcal/mol exothermic.
- The overall thermochemistry is dominated by the potent carbon nucleophile in the reactant, which in turn makes this modification entirely useless for the purposes of a hydrogen-capture system!


The evolution of the dipole moment along the IRC shows very non-linear behaviour (such plots are rarely shown in most published IRC analyses; they should be!), ending of course with the ionic zwitterion that is the imminium borohydride Prod. Indeed the entire reaction coordinate is an unusually vivid example of a non-least motion path!

This simple atom transposition has given us a very instructive exercise in reaction paths, by-passing entirely both Int1 and Int2 (making them hidden intermediates), and converting React → Prod into a concerted reaction. It would be great to probe this convoluted journey using reaction dynamics!
∞Archived as DOI: 10.14469/hpc/3096
‡ Such a species can be seen as a hidden intermediate in the mechanism of reduction of a carboxylic acid by diborane.
†None were shown in the original study.[cite]10.1073/pnas.1709586114[/cite]
Author
Tags: Ammonia borane, animation, Boranes, Chemistry, Cleaning Services, Company: React Group, free energy barrier, Hydroboration, Hydrogen, Matter
As one might infer from the IRC above, the transition state normal vibrational mode has a very unusual form. Given all the bond breaking and making that goes on in this reaction, this mode is in fact almost a pure bond rotation (See DOI: 10.14469/hpc/2705).