Archive for the ‘General’ Category
Tuesday, March 15th, 2022
When you talk π-aromaticity, benzene is the first molecule that springs to mind. But there are smaller molecules that can carry this property; cyclopropenylidene (five atoms) is the smallest in terms of atom count I could think of until now, apart that is from H3+ which is the smallest possible molecule that carries σ-aromaticity. So here I have found what I think is an even smaller aromatic molecule containing only four carbon atoms. And it is not only π-aromatic but σ-aromatic.
(more…)
Posted in General | 5 Comments »
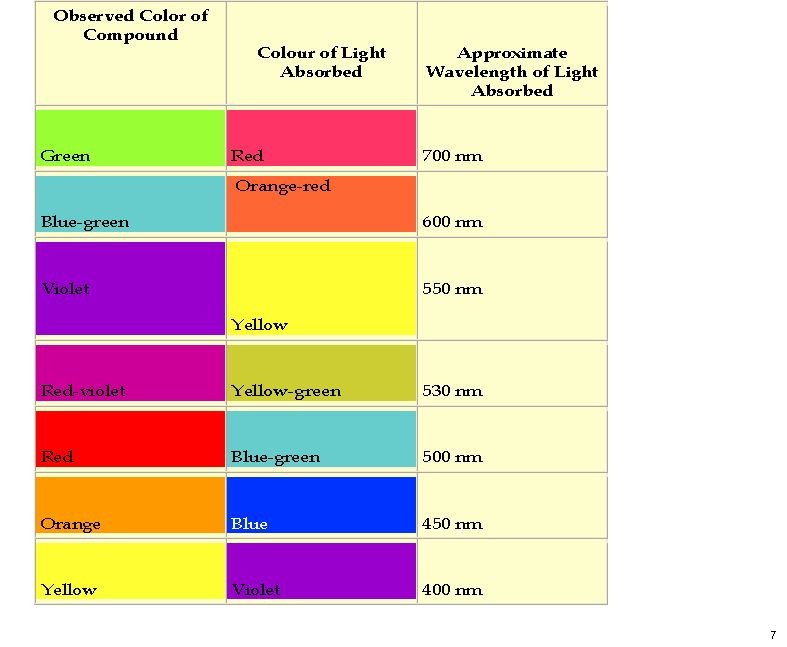
Sunday, February 28th, 2021
I have occasionally covered the topic of colours here, such as those of flowers and minerals, since it is at least possible to illustrate these using photographs or colour charts to illustrate the theme. But when Derek Lowe took a break from his remarkable coverage of the COVID pandemic to highlight a recent article on the active smelling principle in Vetifer oil[1] I could not resist adding a tiny amount to his must-read story.
(more…)
References
- J. Ouyang, H. Bae, S. Jordi, Q.M. Dao, S. Dossenbach, S. Dehn, J.B. Lingnau, C. Kanta De, P. Kraft, and B. List, "The Smelling Principle of Vetiver Oil, Unveiled by Chemical Synthesis", Angewandte Chemie International Edition, vol. 60, pp. 5666-5672, 2021. https://doi.org/10.1002/anie.202014609
Posted in General | 1 Comment »
Saturday, July 25th, 2020
Sometimes a (scientific) thought just pops into one’s mind. Most are probably best not shared with anyone, but since its the summer silly season, I thought I might with this one.
(more…)
Posted in General, Interesting chemistry | 7 Comments »
Sunday, January 14th, 2018
I don’t normally write about the pharmaceutical industry, but I was intrigued by several posts by Derek Lowe (who does cover this area) on the topic of creating new drugs by deuterating existing ones. Thus he covered the first deuterated drug receiving FDA approval last year, having first reviewed the concept back in 2009. So when someone introduced me to sila-haloperidol, I checked to see if Derek had written about it. Apparently not, so here are a few details.
(more…)
Tags:Chemistry, Derek Lowe, Deuterated drug, Drug, drug design, FDA, Food and Drug Administration, Haloperidol, Health, Health/Medical/Pharmaceuticals, HTC HD2 Smartphone, metabolic products, Pharmaceutical industry, Pharmacy, physico-chemical profiles, United States Public Health Service
Posted in General | No Comments »
Thursday, October 5th, 2017
We have heard a lot about OA or Open Access (of journal articles) in the last five years, often in association with the APC (Article Processing Charge) model of funding such OA availability. Rather less discussed is how the model of the peer review of these articles might also evolve into an Open environment. Here I muse about two experiences I had recently.
(more…)
Tags:Academic publishing, article processing charge, author, Company: Facebook, Company: Publons, Company: Twitter, editor, Electronic publishing, Entertainment/Culture, Hybrid open access journal, Internet giants, OA, Open access, Organic Syntheses, Public sphere, Publishing, Scholarly communication, search engines, Social Media & Networking, Technology/Internet
Posted in Chemical IT, General | 5 Comments »
Monday, July 24th, 2017
Bees are having a tough time around the world. Oddly, they are surviving very well in cities. One reason are the wild flower meadows in London and for some summer relief I thought I would tell you the story of the one shown below.
(more…)
Tags:Animal dance, Bee, Behavior, Flower, George, King, Meadow, Person Career, Person Location, Plant reproduction, Pollination, Reproduction, river Brent
Posted in General | 6 Comments »
Friday, November 25th, 2016
Another conference, a Cambridge satellite meeting of OpenCon, and I quote here its mission: “OpenCon is a platform for the next generation to learn about Open Access, Open Education, and Open Data, develop critical skills, and catalyze action toward a more open system of research and education” targeted at students and early career academic professionals. But they do allow a few “late career” professionals to attend as well!
(more…)
Tags:Academia, author, chemist, City: Cambridge, Company: Twitter, ELife, Erin McKiernan, keynote speaker, Max Planck Society, programmer, Simon Deakin, Social Media & Networking, speaker, Technology/Internet, Wellcome Trust
Posted in Chemical IT, General | 3 Comments »
Sunday, September 11th, 2016
To quote from Wikipedia: in chemistry, a carbene is a molecule containing a neutral carbon atom with a valence of two and two unshared valence electrons. The most ubiquitous type of carbene of recent times is the one shown below as 1, often referred to as a resonance stabilised or persistent carbene. This type is of interest because of its ability to act as a ligand to an astonishingly wide variety of metals, with many of the resulting complexes being important catalysts. The Wiki page on persistent carbenes shows them throughout in form 1 below, thus reinforcing the belief that they have a valence of two and by implication six (2×2 shared + 2 unshared) electrons in the valence shell of carbon. Here I consider whether this name is really appropriate.
(more…)
Tags:Carbenes, chemical bonding, energy barrier, free energy, Functional groups, Ligand, Mesoionic carbene, Organometallic chemistry, Persistent carbene, quantum mechanical solution, Reactive intermediates, Transition metal carbene complex, Valence, Valence electron
Posted in crystal_structure_mining, General | 2 Comments »
{kind=link}
{kind=link}